
Haplotype-resolved assembly of a tetraploid potato genome using long reads and low-depth offspring data
Background
基于参考序列的单倍型组装方法 HapTreeand H-PoP
本文使用 hifiasm 组装hifi read 获得一个最初的组装,然后使用193个两个马铃薯品种的后代的低coverage测序数据和一个新的基于k-mer的方法识别四种单倍型,从获得的组装图上组装单倍型。
Results
Overall assembly strategy
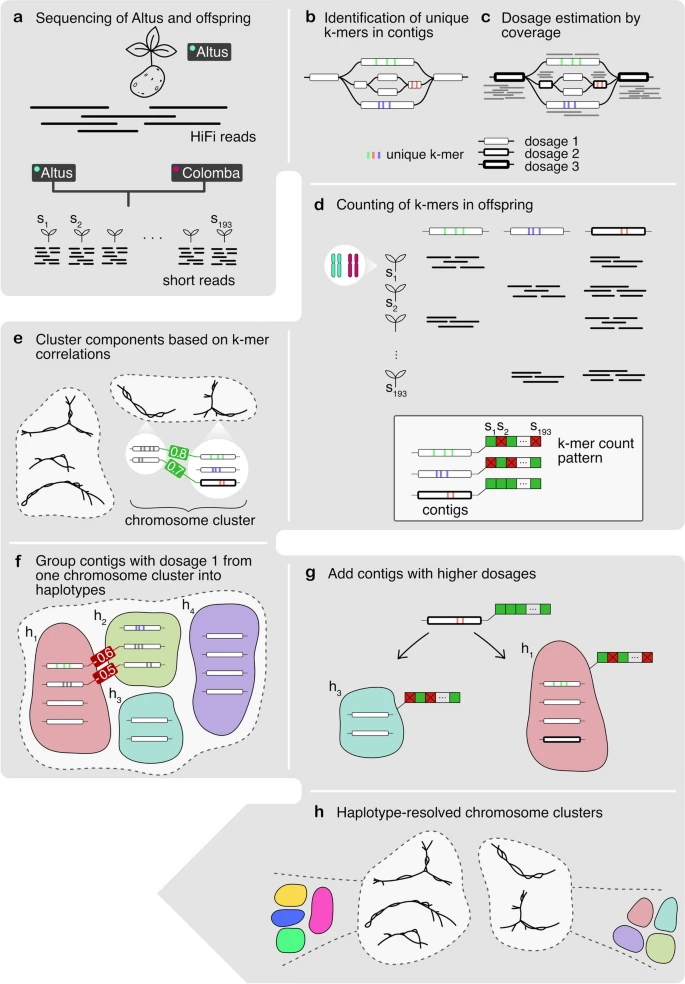
a.获取测序数据, Altus的hifi reads (96x) 以及193个 Altus×Colomba后代的基因组的 short read(6x )。
b.使用 hifiasm 将hifi read 组装成一个初始组装图,其中气泡代表不同的单倍型。对于组装图中的每个的unitig ,检测其独特的k-mers。
c.把hifi read 比对到unitig上,给每个contig估计dosage(1-4,有几个单倍型 )。
d.统计193个后代short reads上独特的k-mer数量,每个unitig由包括193个值的k-mer计数表示,具有相似计数模式的节点很可能在同一单倍型的相近部分。
e.对于组装图中的所有节点,计算 k-mer 计数模式的成对相关性,并对节点在染色体规模聚类。
f.在每个染色体 cluster 中,估计剂量为 1 的节点基于相关性首先被聚类为四个单倍型 cluster。
g.将剂量 > 1 的节点根据 相关性添加到这四个cluster 。
h.对于每条染色体生成 4 个单倍型染色体cluster 。
最后在聚类后的组装图上进行图的遍历完成组装
Initial assembly
每个单倍型进行24×PacBio HiFi 测序,平均coverage = 24x4=96。对103个Altus × Colomba杂交后代 进行 Illumina short-read测序,获得2 × 150 bp paired-end reads ,测序数据中包括两个altus的单倍型,两个colomba的单倍型。平均每个单倍型coverage 为1.5×,平均coverage 为 6 。
用hifiasm组装hifi reads输出了一个包含所有组装的,和未处理的unitigs,部分解决了4个单倍型,变异以泡泡结构在图上表示,既一个unitig分支两个而或多个unitigs
Dosage analysis
dosage (单倍型数量),可以是{1,2,3,4}中的任意值(4倍体)。通过分析比对到unitigs上的reads覆盖深度获得(analyzing the coverage of reads aligned to the unitigs)。
由于hifiasm的图包含overlap,只计算每个节点不与其相邻节点重叠的区域,仅计算这些独特区域的深度作为平均深度。
 b图就是node长度超过100k的覆盖深度图,峰值23,46,69,分别表示dosage 1,2,3。92几乎不可见,可能表示只有很少一部分是纯和区域,并且完全不存在超过100kb的长纯和延伸。
b图就是node长度超过100k的覆盖深度图,峰值23,46,69,分别表示dosage 1,2,3。92几乎不可见,可能表示只有很少一部分是纯和区域,并且完全不存在超过100kb的长纯和延伸。
Analysis of k-mers
统计unitigs中所有可能的 k-mers (k=71) 的数量,确定那些在整个组装图中只出现一次的k-mer,另外丢掉在 Colomba genome 中找到的那些,保证这些 k-mers对 altus 是独一无二的,把这些 k-mer 记录为 unique k-mer,基于unique k-mer 的方法促进了 二倍体后代三倍的单倍型组装和人类88号染色体的挑战性区域的组装。第二个例子中,作者制造了一个独一无二的核苷酸k-mer库,对 long read 编码并组装成 scaffolds。对于每个unitig,使用相应的的 unique k-mer 集合作为node标志,k-mer计数基于Jellyfish。
在后代193个样本中记录 unique k-mer的数量,每个四倍体后代从altus和colomba中分别继承了2个单倍型,通过评估unique k-mer的数量推理一个unitig中继承的copy的数量。对于亲本中dosage 1 ,代表unitig的k-mer在后代基因组可能不存在或存在。对于dosage 2,k-mer可能在后代中不存在,存在一次或存在两次,对于亲本 dosage 3 ,后代必须存在1或者2,对于dosage 4,两个遗传的单倍型都必须源自该unitig。
phase :基因定相,单倍体分型,将多倍体的等位基因,按照亲本正确的定位到父本,母本的染色体,在这里意思是解决单倍型问题。
基于上述,可以将带有unique k-mer的标记为片段有效(phase informative),没有的为片段无效,图c表示node长度分析,一般无效片段都是更短的那些。
Correlation analysis
利用来自同一单倍型的unitigs有相似度k-mer计数,因为相应的单倍型上下文被遗传到后代样本的相同子集。使用 a. 具有有效信息,b. hifi数据和ont数据中有相同dosage估计 (将read比对到unitig上,如果hifi数据中dosage估计是2,ont数据要求是0,1或者2)的unitig 进行分析。在候选unitig 的k-mer计数模式之间计算 Spearman correlation coefficients, 距离相近的基因组位点很可能一起传递给后代,因此由于重组,后代的单倍型信号随着距离的增加而变弱。下图的结果与预期一致。距离小于10Mb的contig之间有强烈的相关性,并随着距离的增加相关性降低。
遗传过程中交叉互换,一个单倍型中相近位置的contig在遗传到后代中更有可能遗传到同一个单倍型中。所以相近位置的contig具有更高的相关性。
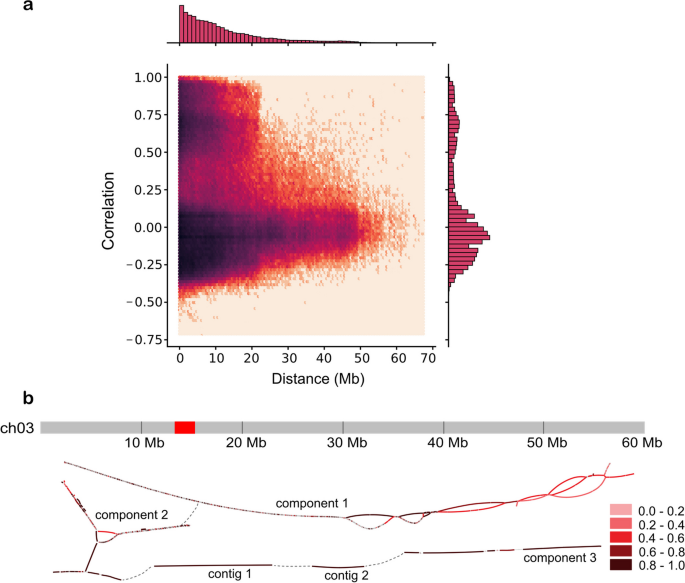
基于高相关性值,可以重新构建出在初始组装中没有连接的区域,像气泡结构和未连接的片段。ch3的重构表示如上图b所示,最初的组装,染色体由3个相连组件和2个更长未连接的contig组成。,通过连接高正相关的contig对,可以重构出染色体的片段结构,并相应的对组件和重叠群排序。
Graph traversal and final haplotype assembly
Method中的方法把每个contig分配给一个单倍型,将cluster拓展到图组件之外,从而匹配属于同一个染色体的组件,导致形成12个假(pesedo)染色体,每个包括4个单倍型,然后,根据组装图中遍历,尽可能的连接聚类后的cluster,生成对应4个单倍型的块(block),称为haplotigs。每个染色体上最长的haplotigs从11.5Mb到34.09Mb,整体的N50是7.54
为了测试使用更少的后代样本能否达到同样的聚类表现,将后代数据下采样到50,100,120,150,发现至少150才能使聚类按照预期工作,50-100不能生成cluster,120输出了5个cluster。这一步我们预期的结果是12 代表12条染色体,第二步,我们才将这些clusters分配到特定的单倍型clusters,在低于150的实验中,即使是12个染色体clusters也没有正确的区分,因此没有采取第二步。只有后代样本数量达到150,k-mer 计数的差异足以区分12个染色体cluster 输出12个 假染色体。后面的分析基于192个后代样本。
思考,如果增加每个后代的coverage 是减少后代个数是否也能生成同样的结果? 实验没有,猜测是因为要重新测序,比较费事费力费钱。
将组装的假染色体和最新的参考序列比对。它的长度 731.3 Mbp(12个单倍体总长度) ,和我们hifi数据中基于k-mer的单倍体基因组估计一致(730Mbp,通过jellyfish统计k-mer计数,然后根据GenomeScope估计基因组大小),每个pesedo染色体(4个单倍型)积累大小是参考染色体的3.5-4倍,总体长度是3.8倍,与先前在其他品种中观察到的某些单倍型结构变异序列丢失一致。
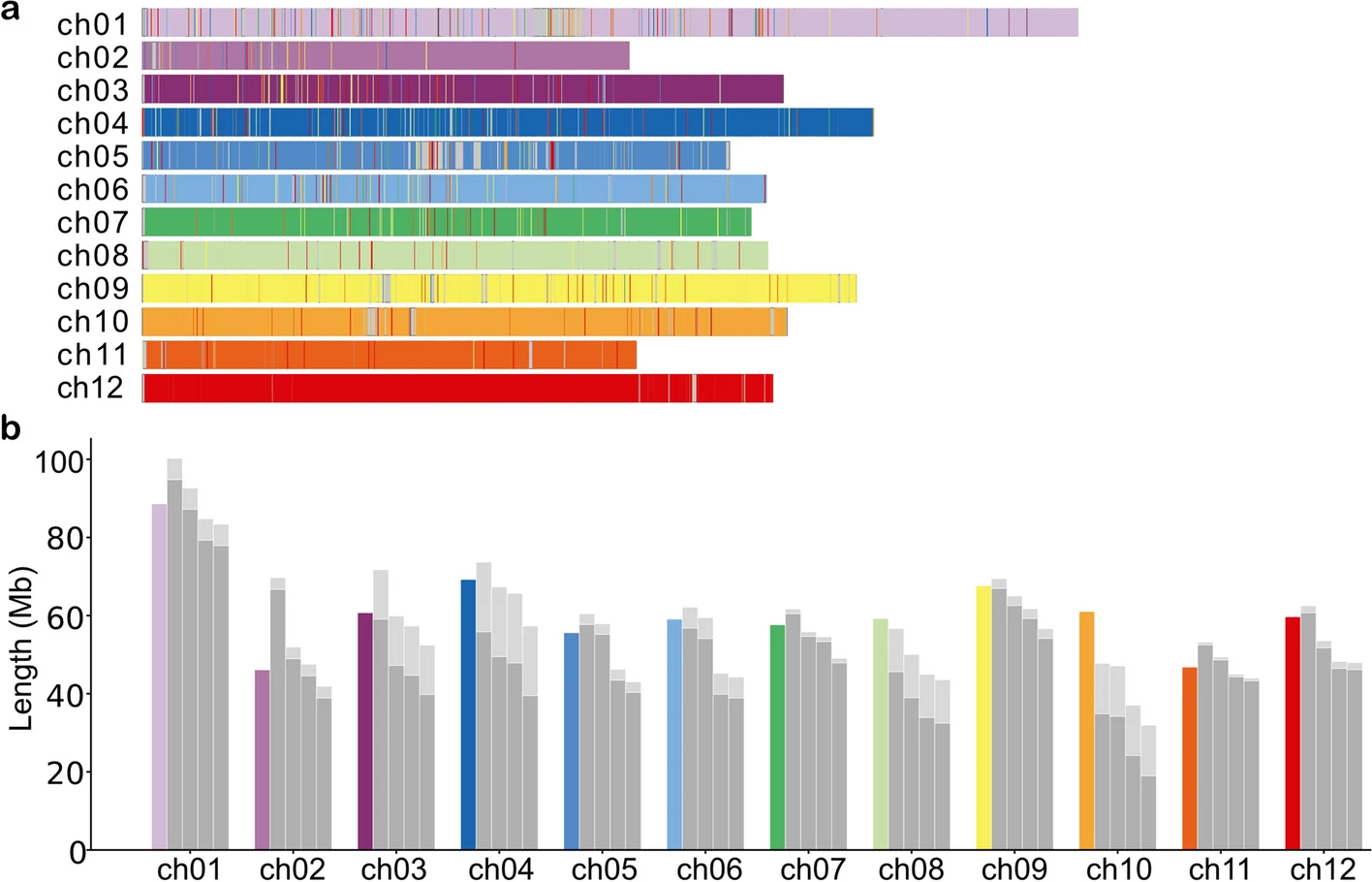 图b中ch07,ch11相当完整,但ch10四个单倍型都明显短于参考序列。每个cluster是上面的浅灰色条表示,第一个步骤中包含在染色体cluster的,但在第二步中必须被排除的。(该值是染色体cluster中为被定为的序列平均分给单个单倍体的长度)。对于ch02 一个单倍型明显比参考序列长,把4个单倍型的cluster比对到参考序列中,这些节点可以特异性的比对到参考序列上,因此不太可能是错误连接,更有可能是更高数量的重复序列。
图b中ch07,ch11相当完整,但ch10四个单倍型都明显短于参考序列。每个cluster是上面的浅灰色条表示,第一个步骤中包含在染色体cluster的,但在第二步中必须被排除的。(该值是染色体cluster中为被定为的序列平均分给单个单倍体的长度)。对于ch02 一个单倍型明显比参考序列长,把4个单倍型的cluster比对到参考序列中,这些节点可以特异性的比对到参考序列上,因此不太可能是错误连接,更有可能是更高数量的重复序列。
图a是把染色体cluster比对到参考序列上的结果,结果表示,组装中所有的序列几乎都是完整的。没有大的gap
Comparison of earlier reference assemblies to reveal structural differences
染色体聚类方法背后的相关信号可以用于检测我们的组装图和以前的参考组装之间的结构差异。这种差异可能表明两个组装中的任何一个都可能存在组装错误,以及所有或某些单倍型的结构差异。在最初的组装图中,我们探测到了图中属于一个component的两个节点集合比对到了不同的染色体上。对于不同染色体上的contig set,这两个set之间的node pair应该几乎没有相关性。实际上,上面讨论的两组,分布形状啊和来自不同的染色体的相关性分布形状非常相似。这可能表示在hifiasm图中存在着错误的连接,本文通过手动进行了纠正。(可以优化的不部分)这样,相关性分析就提供了检测和纠正装配错误的方法。
把本文的组装图和ploid reference Solyntus 比较发现了大量的结构上的差异。例如,本文中染色体8上有两个contig组成的区域可以比对到 Solyntus 参考序列的染色体 1 和 染色体 7 为了确定这个是聚类问题,Solyntus 组装问题,还是两个品种之间的结构差异,对这两个区域进行k-mer计数模式分析。发现这连个区域之间k-mer计数模式和一个染色体内两个部分的计数模式类似,结果表明,要么存在 Altus 和 Solyntus 基因组的大型重新排列,要么存Solyntus 参考基因组的结构是错误的。
Structural analysis
使用 SyntenyPlotteR 和 SyR 进行同源分析,来分析组装中染色体之间的结构差异
使用RagTag 和DMv6.1 参考序列从组装的单倍型中生成scaffolds。然后进行两个两个比对,(h1-h0, h2-h1, h3-h2, h0-DMv6.1,使用minimap2)结果作为输入SyntenyPlotteR
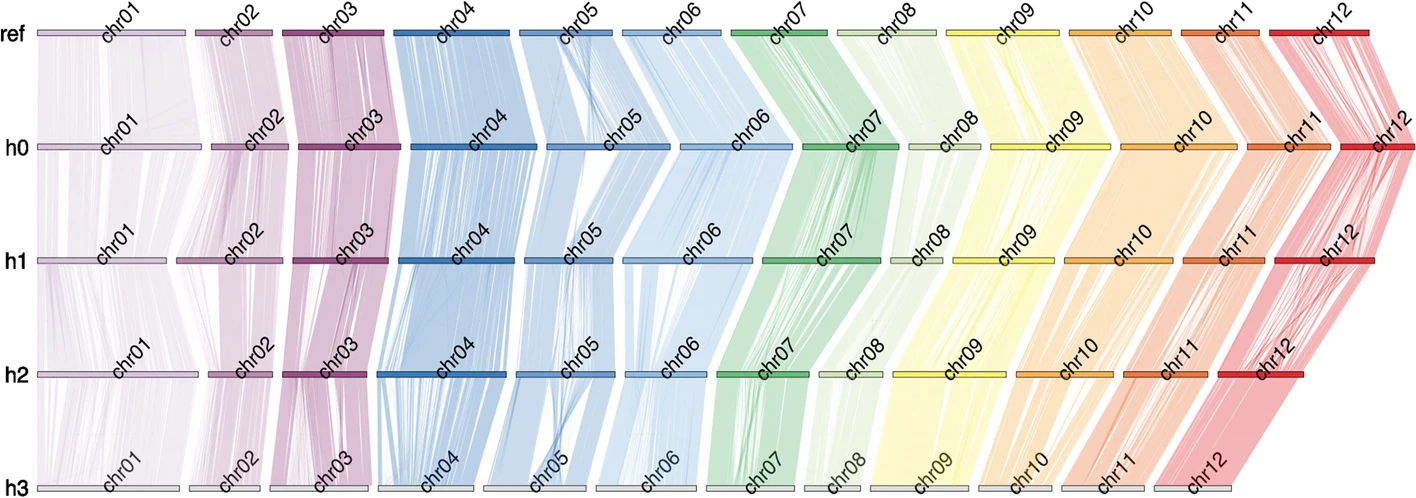
上图提供了单倍型之间及和参考序列的结构差异,值得注意的是,不同染色体之间没有序列片段的易位,这强调了聚类过程的稳健性。
Quality evaluation and comparison to other assemblies
评估组装的质量,运行了更深入的定性和定量分析,并且和额外的土豆组装结果相比,为了评估完整性, 计算 BUSCO scores with BUSCO v5.4.7 using solanales_odb10 获得了完整型分数96.8%,DMv6.1 (97.9%), C88(96.28%), Otava (97.3%)
然后用 RepeatModeler2 and RepeatMas-ker 标记了重复序列 ,最长的重复序列是LTRs(可能是单体较短但是个数很多,构成了长的串联重复)66.81% of repetitive content, 和 Otava 66% DMv6.1 (66.8%) 都类似 最大的重复都是LTRs
使用 Merqury,一个不需要参考序列的组装评估方法,通过比较read set 的 k-mer 和组装 进行 定性评估,使用Illumina data of Altus 用 Meryl 记录k-mer k=21,并且这个结果输入到Merqury 得到 QV value of 45.6677. 更好的 QV value of 51.7 for Otaba。又测试了不同阶段的QV值,对于 phased result ,只包括单倍型中能组装的部分,完整度是89.48%,对于 cluster步骤的结果,被cluster到染色体上,缺乏phased信息,完整度是97.93% . 每个单倍型的 BUSCO scores are 83.1%, 84%, 81.7%, and 81.7%,
为了评估 phasing 的准确性,生成 77Gb of Pore-C data for Altus,然后比对到组装图上,覆盖 node pair 的read的数量设为coverage,评估结果有两个标准,clustered到一个单倍型的node pair (cis pairs)应该倍 Pore-C read支持,不同单倍型的node pair(trans pairs)之间 应该没有支持,影响评估的参数包括,node间的距离,node长度,coverage 。
为了量化分析,重点关注了超过1mb的node pair ,使用不同距离,3,5,7,10Mb,和coverage 从0到70,更高的100-1200,计算相关的真阳和假阳率,画ROC,
对应距离3,5,7,10 Mb的 auc分别是0.96 0.94 0.93 0.93,结果表示Pore-C 有效的支持分型的正确性。
距离设为5 coverage设为30 ,真阳 概率 95.22% ,真阴 概率 92.6% 有251 cis 309 trans 满足这个条件
对于分离较远的位置,即使保持特异性>0.9 Pore-C信号也会随距离边长而下降,总结 Pore-C数据验证了单倍型组装的一致性。
Discussion
Discussion
从头组装方法,该方法使用 hifi long read 和来自后代样本的和 low coverage short read分阶段组装,其单体长度可达染色体臂的长度。设计了一个完整的pipeline,使用单倍型 uniqie的k-mer对染色体进行排序和单倍体分型,以表示自多倍体基因组的组装图。重要的是,这避免了将组装推平为contig的中间步骤,而是直接在图拓扑的上下文中解析单倍型,这可能允许将来添加使用其他数据类型。
我们组装产生的pseudo染色体很好地map到当前的单倍体参考基因组,得到的3.8 倍的序列数据,这表明的单倍型的完整性。通过使用低通量后代测序,我们的方法可以立即在有后代和标准测序设施的育种和研究环境中使用。它不需要单细胞花粉测序技术,这是质量相当的组装体的替代途径。Genome architecture and tetrasomic inheritance of autotetraploid potato 发表了类似的单倍型分型策略,但区别在于它使用自体种群作为后代。因此,我们认为这两种方法是互补的,因为它们可以满足不同的应用设置。
Conclusions
目前组装的缺陷是完整性,在某些染色体上可以改进。这主要是由于两个原因,首先我们用的方法是unique k-mer 对于像土豆这总重复性比较高的基因组挑战很大,第二,测序了 Altus 和 Colomba 的后代,所以必须去除在Colomba 中出现的k-mer 确保只考虑到Altus的信号。
本文方法提供了一种方案,这些数据后代在低覆盖深度或成本限制下确定的。杂交不同品种的做法是常态,如果没有额外的试验工作,通常无法获得自交的广泛数据。
相信本文的方法对马铃薯基因组社区是有用的。
尽管 植物 单倍体分型快速发展,对于复杂的自多倍体基因组来说,单倍型分型 仍有挑战性,
为了解决最难的基因组位点,可以用超长牛津纳米孔技术(ONT)read 与由 PacBio HiFi read 构建的组装图 align。未来可以将本文方法与这些额外的数据类型相结合。目前,由于植物基因组的超长测序读长(> 100 kb)的制备困难,以及我们研究中产生的ONT读长的读长N50s目前较小(N50≤33.43 kb)。但我们预计技术挑战将在未来几年内得到克服。在我们目前基于HiFi的图中,较短的重叠群往往缺乏独特的k-mer,并且12%的基因组是这种重叠群的一部分。将额外的测序数据(例如超长ONT读取)映射到图中可能有助于弥合剩余的差距,从而允许在单倍型序列中包含更多的图节点。
Methods
Data production
Dosage estimation in unitigs
计算每个contig i 去掉和其他contig有overlap部分序列的平均coverage c_i 整体平均coverage(每个单倍型的coverage or 四倍体的coverage/4)是 m,对每个contig估计dosage
c_i > 4.5m 设置 d_i=5 标识这是一个重复的contig
Connection of graph components
将unitigs聚合到单倍型-resolved染色体簇包括两个步骤。首先,尝试在染色体层次解决,染色体可能有多个相连的组件以及额外的单体,确定图中的那些组件属于应该在一起。然后将每个染色体cluster 分成4个不同的clusters,代表4个单倍型。
利用先前计算的k-mer计数(某个node(片段)上的unique k-mer 在193个后代测序数据中的k-mer计数),根据相似k-mer计数将unitigs分组。分组依据同一个单倍型的unitig具有高度相似的k-mer计数模式,而相反模式更有可能来自同一染色体的不同单倍型,模式不相关的unitig可能来自不同的染色体。
两个nodes之间的k-mer计数模式相似性根据seperman correlation coefficient p计算。高正相关代表两个nodes来自同一个单倍型,负相关代表node在不同的单倍型中,只有来自相同染色体会有高的相关性(且距离相近的contig),在两个不同染色体上node,k-mer计数应该是不相关的。
根据最高成对相关性(p>0.5)对所有节点分组,将聚合组件和单个unitigs聚集到染色体cluster里,当发现其中的contigs源自相同的图组件时,合并这些clusters。第一个聚类步骤产生了12个大簇(土豆是4倍体,单倍有12条染色体),定义为相关的染色体cluster或伪染色体。
Clustering of unitigs based on similar k-mer patterns
使用先前计算的doage估计,从dosage1 开始,确定每个染色体的单倍型,因为这些node只能被分到一个组里。首先构建最高相关性种子,然后合并成更大的组添加更多节点。
对每个dosage 为1 的node,创建一个cluster,只包含和它相关性>0.5的unitigs,生成种子cluster,然后根据这些种子cluster之间包含的公共节点合并这些cluster。为了分开不同的组,把高负相关的两个系欸但表示不同的单倍型。
在cluster c_i,c_j 之间如果存在 node pair n_i,n_j\ where\ n_i\in c_i\ and\ n_j \in c_j具有高负相关性( p_{i,j} < -0.3 ) 则构建负边,相反如果存在node pair 其中 p_{i,j} >0.5 则构建正边。如果不存在矛盾边,例如一个从 c_i 到另一个 c_k有正边,但 c_k到 c_j 之间连接有负边,则通过正边连接的两个 c_i 和 c_j cluster可以合并。合并后,和其他节点有高相关性的节点都被分配到cluster里。对于剩余的node,如果3个最好的匹配节点都在 c_i 中,就把它分配到 c_i ,否则就不把它分配到任何一个簇中。
最终,分配具有更高dosage的unitigs到先前计算的单倍型clusters。分配一个dosage x(x\in\{2,3,4\}) 的cluster n ,计算 n 和其他所有cluster中所有nodes的相关性,把node添加到与 n 正相关节点的比率最高的 x个(2,3,4)个cluster
这是分配是保守的,可能有contig未被添加,采取后处理,重新把剩余的节点分配给具有最高相关性的cluster。
Assembly of clustered unitigs
我们从每个染色体的cluster开始,重新构建染色体的contig顺序,去找出它们之间所有的联系,创建更大更连续的单倍型。
如果 phased node 在任一方向只有一个邻居,该邻居也是 phased 的,对于简单气泡结构(源节点,两个分支,汇合节点)。如果源节点,汇合节点是phased的,则两个分支节点中的一个在phased路径上,如果两个分支都不是phased,说明不存在可用的信息选正确的,任选一个,相应的序列被填充为占位符而不是节点序列,表示缺乏正确的单倍型序列信息。
然后考虑剩余部分没有被连接的所有phased节点。这些形成了一个线性的块状结构,对于每个块,我们能够识别两个终端节点,并通过块重新创建节点路径(以及序列,块内部的序列)。为了重建这些单倍型块的顺序,我们随后搜索了仅包含unphased节点的不同块的末端节点之间的路径。对于可以唯一的连接到一个额外块的块,我们将两个块序列连接起来,并再次使用占位符字符来表示中间unphased片段的长度。最后,我们解决了扩展节点路径之间剩余的任何重叠,从而产生了最终的输出序列。
Think
方法好像没用到short read pair 信息,加上可能会提升一些性能