
Deep learning-based prediction of the T cell receptor–antigen binding specificity
部分名词解释
HLA:Human Leukocyte Antigen ,人类白细胞抗原,是人类的主要组织相容性复合体(MHC)的表达产物,该系统是目前所知人体最复杂的多态系统。
MHC:Major Histocompatibility Complex,主要组织相容性复合体,是一组编码动物主要组织相容性抗原的基因群的统称。第一类MHC处理细胞内部产生的蛋白质(例如被病毒感染的细胞产生的病毒蛋白)
CD3:分化簇 3,T 细胞的共受体是一种蛋白质复合物,它由四个不同的链组成,CD3是其中之一。 在哺乳动物中,该复合物含有一个 CD3γ 链,CD3δ 链,和 2 CD3ε 链。 这些链具有被称为一个分子副 T 细胞受体 (TCR)和 ζ-链以产生激活信号的 T 淋巴细胞 。该 TCR,ζ 链和 CD3 分子一起构成的 T 细胞受体复合物。
免疫检查点抑制剂:Immune Checkpoint Inhibitors,通过抑制肿瘤细胞,激活免疫细胞,杀伤肿瘤细胞的药物,包括 CTLA-4 抑制剂与 PD-1、PD-L1 抑制剂等。
新生抗原(Neoantigen)是由肿瘤细胞突变基因编码的新生抗原,是一种由基因点突变、删除突变、基因融合等产生,且与正常细胞表达的蛋白不一样的新的异常蛋白。此类蛋白经过酶解之后形成的多肽片段,作为抗原经过 DC 细胞递呈给 T 细胞,促使 T 细胞变为特异性地识别新生抗原的成熟活化 T 细胞,并使这些活化的 T 细胞增殖。利用新生抗原的免疫活性,可根据肿瘤细胞突变的情况设计合成新生抗原药物,达到治疗的效果。
DC 细胞:树突状细胞,是由加拿大学者 Steinman 于 1973 年发现的,是功能最强的抗原提呈细胞,因其成熟时伸出许多树突样或伪足样突起而得名。
共有的新生抗原:患某些癌症的不同患者间共同存在的突变抗原,而在正常基因组中并不存在。免疫原性高的共有新生抗原有可能筛选用作广谱的治疗性癌症疫苗,用于有相同突变基因的患者。
独有化新生抗原:大多数新生抗原都是独特的突变抗原,在患者与患者之间、肿瘤与肿瘤之间完全不同。因此,独有新生抗原制备药物只能特异性地、一对一地针对每个个体患者或肿瘤。
个人理解
新生抗原是肿瘤细胞基因突变而产生,和正常人体细胞的基因不同,新生抗原在MHC的作用下被T细胞识别,激活免疫系统。新生抗原并不存在于正常的组织细胞中,可以引发真正的肿瘤特异性T细胞免疫反应,不会攻击正常组织细胞。
肿瘤体细胞突变的来源非常广泛,我们必须对这些突变序列进行预测,筛选出最能够特异性引起机体免疫应答反应的候选新生抗原。
预测潜在的肿瘤新生抗原,需要考虑很多因素,包括HLA分型、TCR结合力、MHC亲和力、肿瘤新抗原来源等;还包括用来评估T细胞反应的相关因素,如T细胞识别、TCR分析和免疫细胞分析等
Main
新生抗原(Neoantigens)是主要组织相容性复合体( MHC )呈递在肿瘤细胞表面的上一段短多肽。新生抗原通过与T细胞受体的相互反应作为细胞毒性T细胞(cytotoxic T cells)的目标,在免疫疗法中很关键。
为什么不是所有的新生抗原都能引发T细胞的反应,解释这个是一个基础的且未解决的问题是。关于对T细胞受体(TCR)与MHC结合呈递的新生抗原(二者称为pMHC)的结合特异性的了解就更少了。
这篇文章提出了pMTnet迁移学习模型,可以预测 I 型 pMHCs 和 TCR 的特异性结合
Results
Deep learning TCR–antigen binding specificity
把学习TCR和pMHCs特异性结合分为3个任务
图c 使用netMHCpan模型(主要有LSTM构成)训练得到pMHCs(抗原多肽和MHC结合后)的嵌入向量(embedding)。输入是MHC序列和抗原蛋白质序列,输出是抗原是否能与MHC绑定。此模型预测的绑定概率和真实值的Pearson correlation 达到了0.781。中间的层应该包含pMHC复合物整体结构的重要信息,把最后输出的前一层取出来作为pMHCs的嵌入向量。
图a 使用stacked autoencoder训练一个 TCR序列的嵌入向量 。autoencoder:降维提取特征,模型通过先编码获得一个维度较低的向量然后解码获得原来的向量进行训练
图e 把这两个嵌入向量输入到全连接深度神经网络上去结合TCRs,抗原序列和MHC的知识,最后微调确定TCR和pMHC之间配对的预测模型。采用一种差异学习模式,每个训练周期喂一个正确的TCR-pMHC对,另一个具有相同pMHC的负对。训练后,pMTnet输出0-1的连续变量反向TCR和pMHC的结合强度,所有的结果都针对区别TCR结合特异性,不是抗原和MHC结合。
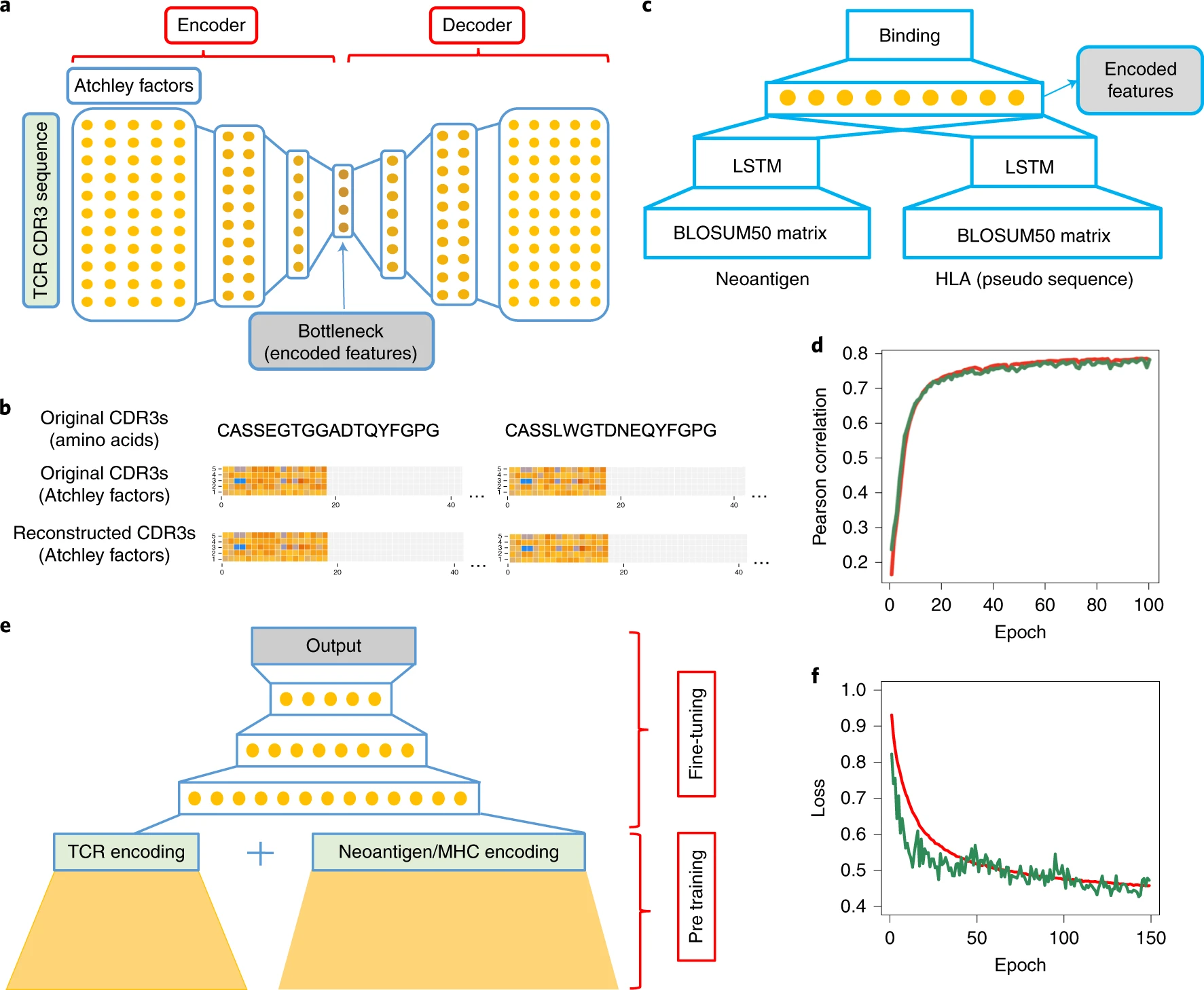
本章节 模型的主要内容估计就这些,下面基本上都是一些实验验证了,内容还挺多的。
pMTnet predicts TCR–pMHC pairing in independent experimental data
说明了TCR-pMHC绑定对是经过实验验证的,训练用的数据集和这些数据是独立的。
用 the area under the curve (AUC) of the receiver operating characteristic (ROC) and precision-recall (PR) 评估。
通过对T细胞的影响验证了TCR-pMHC绑定预测,也就是说,绑定概率(亲和力)越高,T细胞就应该越多克隆倍增。
一些湿实验
Structural analyses support predicted TCR–pMHC interactions
..
Characterizing the TCR–pMHC interactions in human tumours
在肿瘤微环境下研究了几种可能影响T细胞群的抗原。
TCR–neoantigen interactions impact tumour progression and immunotherapy treatment response
..
Discussion
Methods
Embedding TCR CDR3β sequences
通过Atchley factor对 TCR CDR3β 编码,每个氨基酸用5个数值表示,5个数值可以很好的表示每个氨基酸的生化特性。生成的矩阵行数因子数(5),列数为80(序列长度最长为80,猜测,长度不足的填充到80),矩阵经过 stacked autoencoder,具体结构就不说了,最后auto encoder 中最小的全连接层作为 CDR3β 的嵌入向量。
Embedding pMHCs
netMHCpan算法用为序列方法对MHC蛋白编码,伪序列由与多肽接触的氨基酸组成,仅包括34个多态性残基。BLOSUM50矩阵对这34个残基编码,抗原也同样用这个矩阵编码,使用MHC序列而不是类型作为输入,因而可以处理训练集中没有见到过的MHC类型。这篇文章中,把netMHCpan内的MLP换成了LSTM,抗原和MHC的LSTM层输出都是16维,说是可以加快模型收敛。这两个16维向量在同一层组成32维的。然后接一个tanh激活的60个神经元的层,最后接一个1个神经元的层作为输出,训练完后,把60个神经元的全连接层的向量作为pMHC的嵌入向量。
Learning TCR binding specificity of pMHCs
使用迁移学习,利用与预训练好的 TCR 和 pMHCs 编码。固定这些预训练好的模型的参数,然后作为最后的预测模型的前几层。然后 fc300-relu-dropout0.2-fc200-relu-fc100-relu-fc1-tanh 输出。训练过程中,已知的 pMHCs 和 TCRs 相互作用的为阳性,随机错配=它们创建隐形数据(10倍数量)
Differential loss function
他们弄个两个模型,但是共享权重,一个训练阳性数据,一个训练隐形数据(其实就是一个模型,最后的输出层搞两个就行了)
Defining self-antigens
...
Generation of in-house validation data
数据来源
Statistical analyses
写了用了什么软件,什么环境,数据情况,怎么验证的。
最后
这篇文章我猜是生物专业的人弄的,各种实验做的比较充分,尤其临床部分,深度学习方面比较简单,不难理解。