
这篇文章用的数据感觉不错,比较完善可以留着之后用
LongTR is implemented as a C++ package and is accessible on GitHub (https://github.com/gymrek-lab/LongTR) [31]under GNU General Public License v2.0. An archived version of LongTR is available on Zenodo (https://doi.org/10.5281/zenodo.11403979) [32].
Project Adotto Tandem-Repeat Regions and Annotations [33]: https://zenodo.org/record/8329210/files/adotto_repeats.hg38.bed.gz?download=1
PacBio HiFi reads aligned to GRCh38 for HG002, HG003, and HG004 [34]:
https://downloads.pacbcloud.com/public/revio/2022Q4/
PacBio HiFi reads for patients with pathogenic expansions and metadata [34]:
https://downloads.pacbcloud.com/public/dataset/RepeatExpansionDisorders_NoAmp.
HG002 assembly [10]:
https://s3-us-west-2.amazonaws.com/human-pangenomics/T2T/HG002/assemblies/hg002v1.0.1.fasta.gz
adVNTR reference set of repeats [17]:
https://drive.google.com/file/d/1DetpBQySPNe2YAJa4FsjHn9qiRNS3wEV/view
Oxford Nanopore Duplex data for HG002 [19]:
Oxford Nanopore aligned and haplotagged Simplex SUP data for HG002 [35]:
DeepVariant SNP calls used by WhatsHap [34]:
GIAB set of difficult to map regions [21]:
Illumina reads for HG002 [36]:
HipSTR hg38 reference TR set [3]:
https://github.com/HipSTR-Tool/HipSTR-references/raw/master/human/hg38.hipstr_reference.bed.gz
LongTR: genome-wide profiling of genetic variation at tandem repeats from long reads
Abstract
Tandem repeats are ....
Recent improvement in ... improve ...
Here LongTR genotypes tandem repeas from hifi reads
基因分型(Genotyping)是指通过分析和检测个体的DNA序列来确定其基因型的过程。基因型是指某一特定基因或遗传位点上的等位基因组合,通常是指一对染色体上特定基因位置的等位基因的不同版本。
这里的基因分型猜测 :1. 不同的repeat unit 之间的分型。 2. 不同染色体之间的分型 (这种)。还可而已对多个样本进行genotype(多个人的测序, 或者一个人不同位置的测序)
Background
Tandem repeat 的是什么,有什么类型,short tandem repeats (STRs; repeat unit 1–6bp) and variable number tandem repeats (VNTRs; repeat unit 7+ bp), 他是人体基因组突变的重要来源之一,并与越来越多的孟德尔遗传性状和复杂形状有关。然后是本文的提出背景:之前都是用short read做这个工作,但特定的repeat 像高度复杂的TRs 仍然很困难。随着long read 技术的发展,short read 的工具对 long read无效,因为read 长度,碱基准确率,错误类型等方面的区别(additional fig1 展示了两种read错误的区别),所以本文提出适用于long read的工具 LongTR。
Results and discussion
homopolymer 单个重复的碱基,如 AAAAAAAAAAA
partial order alignment 偏序比对,可以获得一种比对的有向无环图。
隐马尔可夫 比对
介绍
LongTR 继承适用short read 的 HipSTR的方法,可以用pacbio或ont 的hifi long read来 Genotype STRs 和 VNTRs。LongTR 吧一个或多个样本的比对信息 和 TRs 的参考集合 作为输入,输出推理的序列和每个locus上每个allele的长度。它使用聚类策略结合partial order alignment 偏序比对,从带有错误的read中推理出单倍型consensus,当read phase信息可用时使用,使用隐马尔可夫模型对序列重新比对,来对每个位置上每个可能的二倍体基因型打分。LongTR支持多样本的调用,采用特定的homopolymer错误模型,输出genotype的质量分数。
实验部分
在来自Adotto 937122 个人类的TRs 参考集合,使用 来自HG002的30x pacbio hifi read 运行。从 解决了的单倍型基因组组装上(使用了多种技术和正交计算方法) 提取等位基因(allele) 作为ground truth 来评估 genotype 的准确性。LongTR输出推理的序列和长度,和等位基因比较。在814319个repeat上,正好提取出来了两个等位基因,有84.7%的序列一致,允许一个碱基误差,有98.5%的长度一致。
为了进一步评估,使用相同的 TRs参考 对犹太人三人组(HG002、HG003 和 HG004) 进行 TR分型,并确定 TR基因型的孟德尔遗传。LongTR ,在参考的等位基因中,至少有一个成员不是纯合的位点中,显示了86%的孟德尔遗传。孟德尔一致性随着基因型质量分组的增加而增加,可用来过滤低质量的调用。homopolymer repeats 有最低的孟德尔一致性 78.3%。 从 HipSTR reference 的840248个 homopolymer repeats(比Adotto有更准确地边界), 训练了一个 pacbio hifi read 的homopolymer error model。 在 Ashkenazi trio 有 83% 和 81% 分别包括和不包括error modeling。使用error model HG002提升了 13% 。
和TRGT(基于PacBio hifi read 的方法,输出推理的序列,和估计的repeat 长度 copy number)对比,在多个方面有更好的性能表现。
然后与 Straglr 和Tandem-genotypes,这两个方法只输出copy number 每个每个locus推理的等位基因序列,使用同样的 reads输入。和Straglr 使用从HipSTR的reference set,对于Tandem-genotypes使用Adotto reference set作为输入。
评估LongTR在 更长的repeat下的能力。和adVNTR比较。adVNTR是一个专门 genotype VNTRs的工具。LongTR只用1.5h ,adVNTR 32h。看他们能否识别大拓展(large expansion 不知道是什么)
评估了LongTR在单独 long read 的能力。使用ONT最新的 Duplex reads(平均长度27kb,pacbio平均15-20k)。使用ONT read 提升了性能。
最后和 short read 的 HipSTR 使用PacBio hifi read 比较。
感觉实验中有较多和实际相关的设置。
Conclusions
在 resolved TR 上提供了准确的结果,和现有工具相比,都更好。 对于大的复杂的插入相关的TR,可能会被参考序列的组装误导。未来使用基于 assembly的方法。可以避免这些限制。
Methods
Overview of the LongTR method
LongTR 继承 HipSTR 去使用 long read genotype STRs 和 VNTRs ,它使用 一个或者多个 samples 的 aligned read 和 预定义的 TRs 的参考坐标集合。 预定义的TRs 可能是简单TRs (e.g., [AC]n),或者多重不同的repeat units (e.g., [AC]n[GT]k[T]l)。对于每个TR,它提取包含它的reads,推理候选TR单倍型,使用隐马尔可夫模型将所有与重复区域有重叠的所有reads 比对到 候选单倍型。最终,输出一个VCF文件,包含每个独立的TR单倍型和相应的质量分数。
Haplotype identification
它首先裁剪目标TR感兴趣的区域的read,区域包括repeat和用户定义的上下文序列窗口(the repeat plus a user-defined window of context sequence)。然后遍历所有裁剪后的序列,包含一个或多个样本中有足够数量reads支持的任意序列作为候选单倍型(最少有两个reads或者多于一个样本中20%的read,或者多余所有样本的5%)。
当repeat长或者带有多个插入删除很复杂的时候,一个单倍型的read很难满足上面的条件,导致它们被排除在集合外了。采用了第二次迭代。对于样本中超过25%的read 缺少相应的表示的单倍型,使用每个这些样本被排除的read形成额外的候选单倍型。这些每个样本之前被排除的reads根据比对到repeat区域的长度排序,使用贪婪的cluster算法形成初始序列cluster。第一个cluster 通过指定第一个序列作为cluster中心形成,对于第二个序列S,计算是否存在cluster中心C,S和C的编辑距离小于T(10),然后通过下面方法修正初始的cluster序列:1.每个cluster的序列使用它上面的reads通过POA生成。这个consensus 更新cluster中心。2.更新完所有cluster中心后,根据中心的长度对cluster排序。3.遍历cluster,合并两个cluster中心编辑距离小于T的cluster,重复此步骤,直到合并完所有可以合并的。5.结束后,只包括reads数量大于 min(10,0.1 x ns)的cluster,ns是每个样本s中被排除的reads数量。最后检查剩余cluster中reads数量和是否大于0.8 x ns,否则增加T(20,50,80,100.。。 700)去放松限制。最后cluster的中心序列添加到潜在的单倍型中去。编辑距离用 Needleman-Wunsch algorithm with parameters gap_score = 1, match_score = 0, mismatch_score = 1.
Alignment of reads to candidate haplotypes
HipSTR 使用严格的比对,将重复序列与候选单倍型比对,假设错误发生在重复单位长度的倍数中,并且在重复中仅发生一次。不适用long read,long read错误可能由于其他程序导致,除了PCR。另外错误可能发生在repeat的不同位置,而不是只有repeat unit size。大部分都是单个碱基的插入删除。为了允许更合理的error model,使用隐马尔可夫模型将reads align到候选单倍型上。
Phasing information
使用HipSTR现有的选项使用read的phase信息准确的识别样本单倍型。考虑phasing 信息,LongTR 每个单倍型至少一个read,unphased reads应该不多于20%总体样本reads
Genotyping
LongTR遍历所有的单倍型对,包括纯合对,对每个可能的genotype 使用所有观测到的read计算分数。
Genome-wide profiling of heritable and de novo STR variations
Abstract
genotyping and phasing STRs from Illumina sequencing data
Main
Methods
The HipSTR algorithm.
Modeling PCR stutter.
PCR stutter(PCR 滞后)是指在聚合酶链式反应(PCR)过程中,特别是在扩增短串联重复序列(STRs)或微卫星序列时,出现的非预期 DNA 产物。具体来说,PCR stutter 是由于 DNA 聚合酶在复制重复序列时发生滑动,导致扩增产物中出现比目标序列略短或略长的额外条带或峰。
HiFi测序的一个显著特点是它在直接读取长DNA片段时,通常不需要或仅需要少量的PCR步骤。这减少了PCR扩增引入的错误,包括PCR stutter。
Generating candidate alleles.
识别最初的STR等位基因集合。HipSTR选择能横跨STR的reads,他要求read的两端和参考基因组至少匹配10bp,并且两端都不能于比对上有或者下游的参考基因组有比15bp更长的精确匹配。(It requires that both ends of a read match the reference genome for at least 10 bp, and that neither end of the read has a longer exact match with the reference genome 15 bp upstream or downstream from its alignment ??)。
如果一个序列出现在2个或更多的reads,并且超过20%的样本read数量,HipSTR把它作为候选allele。
HipSTR使用迭代方法识别新候选allele。在每轮genotype结束后,他计算每个样本的最可能的genotype,并且将 在样本的genotype和最可能的allele相关的所有reads重新比对,每个alignment 在STR区域生成一个序列。如果在一个样本里,有两个或多个alignments相同,HipSTR选择这个序列作为新的候选allele。
这个是在所有处理过程结束后,重新再来一遍。。或者再来多遍。。。
Computing genotype likelihoods
genotype 可能性模型集合了每个read的phase可能性和alignment可能性的信息。
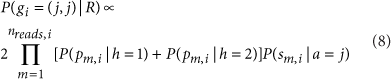
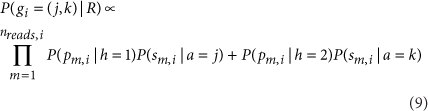
Read phasing likelihoods
计算每个read的phasing 可能性,
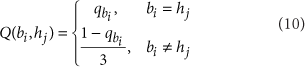 将read上的每个比对到snp hj 上的碱基计算Q,并把Q值相乘得到这个read的phasing 可能性。实际上,SNP在STR区域可能是错误的,所以排除STR区域周围15bp的SNP。如果没有phased SNP信息可用,HipSTR对两条链设置相同的phasing 可能性
将read上的每个比对到snp hj 上的碱基计算Q,并把Q值相乘得到这个read的phasing 可能性。实际上,SNP在STR区域可能是错误的,所以排除STR区域周围15bp的SNP。如果没有phased SNP信息可用,HipSTR对两条链设置相同的phasing 可能性
Read alignment likelihoods
使用了两个分别的模型计算可能性,计算read align到 flanking regions 的可能性 和 到 STR sequences的可能性。
Aligning reads to flanking sequences 使用隐马尔可夫模型。使用了三个矩阵递归计算碱基b1-bi和h1-hj的最大对数似然。结合 t x->y 的值I(从隐藏状态x到隐藏状态y的对数概率),填充每个矩阵。
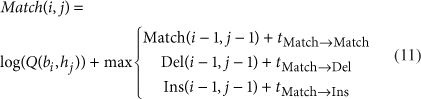
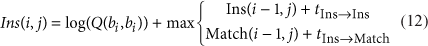
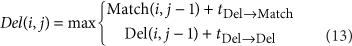
Aligning reads to STR sequence. 好多处理都是为了避免PCR stutter的影响。