
负二项分布描述的是第r次成功之前失败的次数。
负二项分布与二项分布不一样,它表示在一些列伯努利试验中,成功概率为 p,成功次数为 r之前的失败次数的概率分布。表示成:
Abstract
Mettagenomic Hi-C 作用,分类,相关方法局限性,本文方法MetaC的框架,优势和效果。
作用 可以识别同一物理细胞内contigs之间的关系。
分类 short 和long metaHi-C 数据。
相关方法局限性 只能用short metaHi-C数据并且有很大提升空间
Introduction
宏基因组学介绍。
Metagenomic Hi-C technique (metaHi-C) 技术介绍,作用,缺陷,简要介绍方法的流程,和几个现有方法。
然后说了现有方法的缺陷和原因,所以需要本文。。。
介绍本文的方法,主要流程,特点,相对于其他方法的优势,及部分实验结果的简要说明。
在不需要分离环境中的微生物的情况下研究群落结构和生态系统中的代理。新技术 Hi-C 为单样本的物种多样性和微生物之间的相互作用提供了新 insight。
Metagenomic Hi-C technique (metaHi-C) 在同一微生物样本内进行,在同一物理细胞的 loci之间创建dna-dna连接,生成大大量的paired-end Hi-C short reads。 reads组装成contigs后,Hi-C reads比对到这些contigs上,因此,连接到的两个contig 的Hi-C reads的数量,反映了contig-contig之间关系的临近性。原始的Hi-C数据会受到系统偏差的影响,需要正常(纠错?)化原始数据。他们之前提出了3中系统偏差,包括,重叠群上的酶促限制位点数量、重叠群长度和重叠群覆盖率 ,提出了HiCzin纠错。纠错后,可以使用Hi-C连接将片段的contigs分成metagenome-assembled genomes(MAGs)组。这个过程是Hi-C bin,可以构建宏基因组组装的大概,现有的方法有MetaTor,bin3C 和HiCBin。
对于现在的这些计算方法,有很大的提升空间。Knight-Ruiz使用bin3C,当raw Hi-C矩阵高度稀疏的时候,可能无法生成双随机矩阵。MetaTOR在binning操作中使用经典Newman-Girvan modularity函数,在复杂hic联系网络中,可能不能识别小的genome。hiczin和hicbin在估计contig丰富度(覆盖深度)和生成contig标记时,需要大量计算资源,这个指的是将核苷酸序列分配到不同的分类水平。另外,hiczin和hicbin使用TAXAassign通过运行blast将序列比对到精选的核苷酸参考数据库,对contig在species(种类)层次标记。另外,现有的计算方法都是在short-read metahic数据上设计的但很少有benchmark。随着三代数据的发展,有些实验开始使用nanopore和pacbio生成long-read metahic数据。然而现有的在short read metahic上的方法,当用来处理long read metahic时有一些难困难。hicbin在short read metahic上表现很好,但在long read metahic上急剧恶化,原因是采用了归一化的hiczin方法,只有一小部分组装的contig可以通过taxaassign在物种水平上成功标记。另外,long reads 组装的contig 使用taxaassign标记种类具有挑战性,所以限制了hicbin和hiczin在long read metahic上的有效性。因此需要新计算方法填充这些空白。
本文metacc,一个可拓展的集成框架,可以同时适合long read 和short read metahic数据集。框架中,normcc对原始的metagenomoic hic 连接进行标准化(纠错)。相对于hiczin依赖于估计contig丰富度,nomcc使用负二项式回归模型表示contig丰富度,这个模型基于重叠群上的限制性位点数量、重叠群长度以及重叠群内邻近连接事件的数量。所以normcc不需要contig丰富度的估计。hiczin需要contig 注释,相反,normcc使用负二项式回归对每个contig的相邻建模,不需要contig注释。使用和成酵母数据集,验证normcc各方面比hiczin要好。利用了nomcc可以获得高质量MAGs,比任何现有binning tools都好。
Methods
Overview of MetaCC
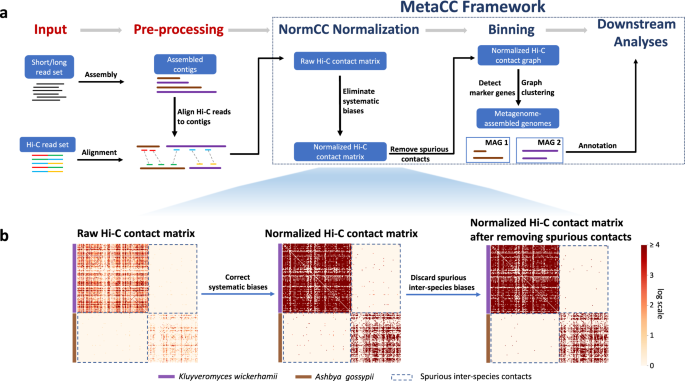
metacc是一个综合的分析框架,对于short read 和long read metahic数据集都适用,主要包含4个部分。
normcc,可拓展的有效的normalization方法,从原始 的metagenomic hic 联系矩阵中估计系统偏差。
丢弃来自不同物种contig 的虚假hic 连接
基于归一化hic连接图,使用 Leiden clustering 方法检索高质量MAGs,自动调整超参数。
通过集中计算策略,表征微生物生态系统的结构。
Real metaHi-C datasets
用了两个公共metagenomic hic dataset,包括两个 short read metahic 数据集和两个long read metahic数据集。两个short read metahic 分别来自人体肠道和废水。hic数据通过限制性内切酶Sau3AI 和MluCI,所有read和hic数据都是用Illumina platforms 长度 150 bp。
两个 long read metahic 来自牛瘤胃和羊肠道。牛瘤胃的数据由pacbio 未纠错的read和hic libraries组成。hic通过限制性内切酶Sau3AI 和MluCI 在Illumina HiSeq 2000 以80bp长度测序。羊肠道的long read是pacbio hifi read。hic数据是150bp长度。
Data processing
对于metagenomic hic实验,清理read步骤非常重要,因为由于adaptor 序列,低质量read,和pcr重复会导致严重的问题。因此对hic read libraries使用标准的清理程序,使用了来自BBTools suite 的 bbduk 进行。
short read 数据,使用MEGAHIT组装。pacbio的两个long read metahic 数据集用原论文提供的组装contig。long read用canu组装,用Pilon和illumina数据polish 两次。hifi reads有一个更新后的版本是用metaflye组装的。
最后,将处理后的paired end hic read 比对到assembled contig上。使用bwa-mem比对。移除没有比对上的read,次要(secondnary)比对,补充比对(supplementary alignments)和低质量比对。(length<30 or score <30)。原始contig-contig 联系通过记录比对到两个contig的hic read pair 数量获得,反映了两个contig 之间的联系。同样定义了比对到同一个contig的hic read pair。因为越短的contig的hic 信号越少,并且且限制性位点会导致更高的变异,削弱了下游分析的稳定性,所以限制最小contig length(1000bp),限制位点数量(1),最小hic 联系数量(2个比对到不同contig的hic read pair,一个比对到同一个contig的pair)去过滤有问题的contig。然后根据hic pair-end read比对生成原始hic contact矩阵。对角线和非对角线分别代表contig内和contig间的hic连接。hic是为了探索contig间的连接,所以之后hic contact表示contig间的连接。
NormCC normalization module in MetaCC
normcc 是一个可拓展的有效的归一化模型,可以有效的消除限制性位点,contig长度,原始hic contacts的coverage 的偏差。
定义H标记原始hic contact 矩阵。Mi为contig i 和其他所有contig的 hic 信号连接总数。(hic contact矩阵的第i行之和,不包括对角线上的值)
对M_i 用负二项式分布建模。
\theta 是负二项分布的参数,平均值\mu_i 取决于原始metagenomic hic contact的3个系统偏差,contig中限制位点数量s_i,contig长度l_i和coverage c_i。用log函数对u_i 基于这三个偏差建模。
酶限制位点和contig长度可以直接确定,获取覆盖深度很耗时,设计了一个静态模型,使用已知的元素获取位置的coverage。 N_i 表示contig i 相邻连接事件的数量(hic 数据在contig i 上的覆盖深度,hic within coverage)。
基于上面你的公式,开发了第一个负二项回归模型NBR_1,系统偏差和作为预测变量参数,within hic contact 作为响应变量。NBR_i的残差可以写为 N_i / v_i。
假设只有这三个变量会对同一个contig内部的近似临近事件有主要影响。通过残差,消除了 within contig hic contacts的内部影响,随意残差可以表示为
C是一个常量。除了上面说的三个参数外,不同重叠群之间的临近成都也会影响他们之间临近连接时间的数量。所以within contig 的假设不适用于across cotig hic contact。
通过上面的公式,可以获得contig coverage
所以,遂于未知独立变量c_i 可以同上面的公式表示,但有些参数是未解决的。把c_i 带入 log(u_i)的公式得
基于公式(2)和(10),获得第二个负二项分布NBR_2,在NBR_2中,M_i 作为相应变量,s_i, l_i, N_i 作为预测变量,获得给定contig i 的平均分布 u_i。 估计的u_i 可以通过如下获得,\hat 对应的是相应的最大似然估计
另外,u_i 代表的是估计的临近连接时间数量平均值,这个估计仅与那三个因素有关,话句话说,u_i 表示的是contig i 和其他contig 临近的能力。为了解决这些偏差对contig之间生成hic 交互的能里的变化。标准化contig i 和contig j之间的原始hic contacts, 把他们除以ui*uj的平方根
C是一个缩放常量。用来调整hic contact 标准化后的范围,避免他们太小,uiuj的平方根可以表示跨contig的contact数量的缩放几何平均值。基于三个偏差因子预测,基于假设,实际的跨contig的hic contact 和只考虑这三个因素之间的偏差主要是由于空间临近性,因此可以反应跨contig的无偏临近性。
Discarding spurious inter-species contacts based on NormCC-normalized Hi-C contacts
虚假的种间contact指的是,由于实验噪声产生的来自不同genomes之间的重叠群之间的contact。根据经验将normcc 标准化后 contact 低于p%(5%)的是为虚假丢掉。
Genome binning in MetaCC
在用normcc修正系统偏差和去除假hic contact 后,处理后的hic contact矩阵变成一个没有自循环的带权图,标准是任意两个contig之间的normalized hic contacts。利用Leiden graph clustering 将图cluster到草稿bins 中。leiden是一个模块化的群体探测算法,采用残心策略优化模块化算法。原始的Newman-Girvan模块会受到分辨率的限制,可能识别不到小bins,本文利用基于reichardt and bornholdt’s pott model
e_ij是contig i 和contig j 间的边权重,d_i 和 d_j 表示contig i 和contig j的度。n是图中边的数量 r表示一个分辨率参数,Delta_ij 是一个只是函数,如果contig i,j 属于同一个群值为1。参数 r 可以表示为可以看作配置空的部分和群内部的连接之间的相对重要性,r越多往往会产生更多的群。
用FragGeneScan和HMMER 检测组装的contig 中的 single-copy marker genes 估计metagenomic 中的genomes 的数量,记为k。设置最小bin size 150kbp,稍小于最小的细菌染色体的长度,只考虑大于这个长度的作为resolved MAGs。然后,目标是,选择一个合适的r使MAGs数量和估计的样本中genome的数量对齐。测试了从小到大一系列 r 的值,对每个 r ,记录MAGs 的数量 k_r。 考虑到可能不会在特定的种类中探测到marker,导致的潜在少估计了genome的数量,r被确定为 第一个得到的MAGs的数量超过估计的genomes数量时,对应的值。
确定好参数r后,将cotig 聚类到MAGs,获得初始的MetaCC binning 结果。
Evaluating the quality of recovered MAGs
用CheckM评估获得的MAGs。将 完整性completeness >50% contamiantion <= 10% 的作为高质量MAGs,高质量的细分成三类
near-complete: com>=90%,con<=10%
subtantially complete: com>=70%,con<=10%
moderately complete: com>=50%,con<=10%
Cleaning partially contaminated bins in MetaCC
除了高质量MAGs,仍存在部分被污染的完整性高于50%的bins,在最初的bins里污染高于10%。类似于其他binner,MetaTor 和 HiCBin,本文选出并清理所有部分被污染的bins,通过使用Leiden 算法划分被污染的bins中的contigs。参数设为1,因为被部分污染的bins内的组数很小。处理后的结果会产生更小的bins,记为sub-bins,保留那些尺寸大于最小限制(150kbp)的bins,将他们合并到最初的bin set,获取metacc binning 的最终bin set。
Assessing the performance of normalization and spurious contact removal on a synthetic yeast metaHi-C dataset
评估 normalization 标准化 和 虚假 contact 连接的性能,在一个合成的酵母样本中,样本包含13个酵母物种。原始的shotgun有85.7 百万个101bp的read pair , hic 库包含81百万个 100bp 的read pair。read清理,contig组装和hic read alignment 步骤和 shot read metahic 数据及的处理一样。因为物种是已知的,可以确定assembled contig的物种,所以intra-species hic contact 和虚假的inter-species hic contact 都可以进行 benchmarking 分析。
Estimating the coverages of assembled contigs
short read 比对到组装的contig上估计coverage,用BBMap 和minimap2 分别比对short reads 和long reads,用samtools转化成bam文件然后程序计算。
MAG analyses on the human gut short-read metaHi-C dataset
因为人体肠道的很多细菌在之前的研究中已经被识别了,我们可以根据存储库中已知的肠道细菌,评估从 short read metahic dataset 获得bins。Unified Human Gastrointestinal Genome (UHGG) 是最大的公共肠道微生物参考数据库。用mash(10000 sketche per genome)计算UHGG 物种级别表征和获得的bins之间的mash distance。mash distance 可作为1-ANI ( average nucleotide identity ,平均核苷酸一致性,两个序列之间的相似度) 的可靠代理,一般用95% ANI 作为物种边界,对应mash 的 0.05。所以,如果bin和参考基因组中的序列的mash distance <0.5,把这个bin分配给这个物种。如果一个bin被分配给多个物种,认为它是嵌合(chimeric )的。
MAG analyses on two long-read metaHi-C datasets
为识别那些metacc binning和其他基于hic的binners的结果中接近完全的bins之间的重叠bins。用mash with 10000 sketches per bin 计算两个bin sets 中bin 的距离。距离小于0.01的认为是同一个genome 的MAGs。此外,评估不同binner 获取物种多样性的能力,用GTDB-TK 注释高质量bins去获取不同binner的高质量MAGs的分类信息。
Plasmid analyses on the sheep gut long-read metaHi-C dataset
metacc binning 获得的709个高质量MAGs 中共6320个contigs,先通过PPR-Meta用cut-off 0.5去识别潜在的plasmid contig。这个方法识别到了111(1.7%)个潜在的plasmid contigs。然后进一步通过Platon 用Sensitivity 模式排除潜在的染色体contigs。最终,99个contigs最终被识别为高置信 plasmids。用BLAST查询这99个plasmid contigs,与和reference plasmid genomes from NCBI至少有1000bp的contig超过95%。
Quality control of all metaHi-C datasets
qc3C in k-mer 模式和默认参数,评估不同metahic 数据的hic质量。
Other algorithms used in benchmarking
HiCzin,VAMB, MetaTOR,bin3C,HiCBin 都是用默认参数。
Results
NormCC comprehensively corrected all systematic biases existing in a synthetic yeast metaHi-C dataset
计算了intra-species contact 和相应contig pairs 的限制位点,长度,coverage间的Pearson correlation coefficients (相关系数,线性值为1,非线性为0),分别为0.429,0.400,0.184,表现出关于这三个因素在hic-contact上强的偏差。normcc 标准化后,系数降为0.094,0.090,0.004。表示normcc能有效修正系统偏差。
NormCC outperformed HiCzin on the spurious contact removal, contig clustering, and computational time
各个方面和hiczin比较,说明比它结果好。
MetaCC binning achieved the best performance of MAG retrieval on short-read metaHi-C datasets
CheckM评估结果。
MetaCC binning markedly outperformed existing binners on long-read metaHi-C datasets
MetaCC binning identified and expanded the order Erysipelotrichales from the cow rumen and sheep gut samples
根据GTDB-TK的标记,8个高质量MAGs 属于Erysipelotrichales order(目?)。 MetaTor,bin3C,HiCBin的结果中分别是 5,3,1。8个bins中3 个可以标注为种类级别,表示还有潜力拓展这个目的种类。
Plasmid analyses among high-quality MAGs retrieved by MetaCC binning from the sheep gut sample
基于shotgun的binning方法通常不存在多个拷贝的plasmid,因为这类方法使用contig 的coverage进行binning。提取了MAGs中 coverage>2* mean coverage of their 的plasmid contig。两个contig分别是3x 和 5x。另一个contig 和NCBI plasmid reference genome 有10166bp的alignment 长度(98.4)。这个contig属于的bin被GTDB-TK注释为大肠杆菌。
Running time of the overall MetaCC pipeline
2.4Ghz e5-2665 50G 内存,在人类肠道short read,废水short read, 牛胃long read,羊肠 long read 数据集,分别用了19分钟,56分钟和109分钟,