
每日小结
-
2021-03-31
完成
看了点纠错知识
服务器跑商三代数据 align,sort,merge
明日计划
三代数据比对结果(3970000-39870000)
二代比对结果到(3970000-39870000)上深度信息有点奇怪等二代数据比对到chr13结果出来后再看看,
看看三代自纠错和二代数据纠三代 -
2021-04-02
- 三代数据比对到chr13中完成,结果也是比较怪,有部分长度的序列深度特别高,结果在2_chr13_pacbio_align.sort.bam中
- 二代的在服务器上出了点问题
- 放假前把二代数据比对到全基因组上,看看效果
- 之后应该看纠错的知识了
-
2021-04-05
- 将实验室电脑装成ubuntu系统,并配置相应的实验环境。
- 看了看比对到全基因组上的结果,发现2.JY中是两个拷贝,不是给的4个拷贝,对于不同的拷贝,其只是有偶尔一个碱基不同
-
2021-04-06
- 得到三代数据的比对结果后,发现拷贝位置的深度确实是其他位置的四倍,重新查看二代数据的比对信息时,发现比对的二代数据是选择错误的H25,重新比对二代数据
-
2021-04-07
- 获得二代数据的比对结果,使用IGV软件进行查看,发现,二代数据比对深度不平稳,但也明显可以看出是其他正常区域的四倍,三代数据深度平稳,是其他位置的四倍,其中四个拷贝,不同的地方仅仅是一个碱基,考虑简单的排列组合进行组装是否可行,以及此项目是否有继续进行的必要。
- 部分拷贝区域内的二代数据深度低的部分,三代数据中的错误率也较高,但并不绝对,这一点不仅仅在多个拷贝的区域这样,具体原因仍需要考虑(组装的参考基因组的问题?)
- 几个拷贝之间基本上相同,主要应该解决的问题是否是几个拷贝间的连接问题。以及不同拷贝中不同部分的顺序
- 明天看看两个拷贝接触位置的覆盖深度,从而可以确定,拷贝之间是否是相连的。以及确定三代数据的长度范围。
-
2021-04-08
- 三代数据的长度从 几k 到 一百k 左右都存在。查看了拷贝接触位置的覆盖深度,没有什么结论,疑问:最后一个拷贝是否不是和之前的拷贝的长度相同,也就是最后一个拷贝的中间位置连接之后的部分
-
2021-04-09
- 开会 仔细注意两边连接处的详细信息,
- 将74k拷贝后接的于它开始相同的到nnnnnnn之间的碱基去掉,进行全基因组的比对,查看比对结果,
- 多个拷贝连接失败的原因,假设拷贝是-AXAXAXAX-,AX是一个拷贝,第一个X后边接A的信号远强于接-的信号,但是接A就会形成的一个环,然后无限循环无法停住,所以原有的所有组装软件都拼接不起来。其次,最后一个X也并非和前面几个X的长度相同。
- 下一步,查看删除后的比对结果,然后对三代数据进行纠错,提取出所有于三代数据有相关的reads,设计算法进行组装
-
2021-04-12
- 早上查看比对结果时,发现使用minimap2比对三代数据时意外终止,猜测原因可能是自己使用多线程导致内存不够用,或者代码对多线程有bug,原来使用14个线程跑,使用8个线程时恢复正常,继续跑三代数据比对
- 下午去健身了~~~
- 晚上回到实验室查看比对结果,发现有一条约3k的read比对到了拷贝,以及拷贝后的序列,中间有约1k的deletion,将这条read添加到参考基因组中重新比对,虽然只有一条read偶然性较大,若比对结果理想,仍能表明这个课题基本上能有个好的结果,这可以算是一个重大进展了。
- 第一次体会到了科研的乐趣,看到这条read时,心中有股难以言喻的喜悦之情,跑出去坐了10分钟才平息下来,哈哈。
-
2021-04-13
- 查看了比对结果,coverage只有二十几,并不是很完美,但感觉也够用了,
- 重新修改了一个参考基因组,重新比对看看结果会不会好一点
- 修正三代数据
- 开始看sga源码,论文
-
2021-04-14
- coverage还是只有20多,而且二代数据的coverage很多位置是0,猜测原因开始是添加的三代read中错误率较高,可以等之后纠错后再找到read添加测试
- 纠错,开始时发现软件经常意外终止,开始还以为是软件有bug,后面才知道是自己内存不够大,而且swap分区也比较小,把swap分区调大(64G)后就可以了,之前minimap2的意外终止也可能是这个原因
-
2021-04-15
- ubuntu出问题了,进不去桌面,命令行正常,重装了一遍显卡的驱动后就好了,自带的显卡驱动也太辣鸡了呀
- 查看纠错后的read,输入的read数量少了很多。。。
- 手动将纠错之后的read添加到参考基因组上进行比对,
-
2021-04-16
- 手动将纠错之后的read添加到参考基因组上进行比对后,在三代数据中,添加的和拷贝的连接处有一段的coverage比较高,有500左右,暂不清楚是什么原因
- 将原始的参考数据中的断开的区域中的nnnn去掉后,在进行比对,查看是否能有其他的数据能够连接上这两段
- 进行了三代数据的纠错,不知道两天多能不能跑完
- 对两个拷贝的进行的比对
- 继续看组装算法的论文
-
2021-04-19
- coverage到达500左右,可能是因为进行了纠错的缘故,纠错后的序列 更靠近的了其他序列
- 还有只有那个read能连接起来两端序列,但第一段后还是又可以比对上的序列的
- 要完成三代数据纠错至少要十几天,暂时放弃,之后应该会自己在组装的过程中实现纠错
- 进行了4.JY的拷贝位置的比对,以及2.H25的全基因比对和删掉拷贝后边的重复的全基因比对
-
2021-04-20
- 2.H25中并没有找到能够连接到后面的read,初步猜想是终端的间隔有点长,导致基因组不好比对上
- 在拷贝位置的比对结果中,发现有某几个区域的coverage特别高,其他的正常,可能这个位置是某个基因,在其他的地方也有相同的
-
2021-04-21
- 论文论文,后面在开始写代码之前估计就时一直看论文了。。
-
2021-04-28
- 开始写组装算法。
资料
生物信息学博客
- HOMOLOG.US - FRONTIER IN BIOINFORMATICS
- DE BRUIJN GRAPHS - I
- DE BRUIJN GRAPHS - II
- DE BRUIJN GRAPHS - III
- STRING GRAPH ASSEMBLER
- STRING GRAPH OF A GENOME
论文
- Long-read error correction: a survey and qualitative comparison
- The fragment assembly string graph
- Efficient construction of an assembly string graph using the FM-index
- Efficient de novo assembly of large genomesusing compressed data structures
- Fast and accurate short read alignment with Burrows–Wheeler transform
- Minimap2: pairwise alignment for nucleotidesequences
- Fast and accurate long-read assembly with wtdbg2
- Canu: scalable and accurate long-read assembly viaadaptivek-mer weighting and repeat separation
知识点
-
mapping:只提供read比对的参考序列上的位置信息;alignment:不仅提供位置信息,还有他们的实际位点和参考位点之间的匹配、不匹配、插入或删除的关系
-
- global alignment :需要比对整个查询序列,查询序列在参考序列上的比对结果可能会不连续,一般用来比较同源基因,或者基因的差异性
- local alignment :查找最能匹配上参考序列的局部区域,比对上的结果可能是查询序列的子序列,
Fast and accurate short read alignment with Burrows–Wheelertransform
prefix trie
the prefix trie of googol
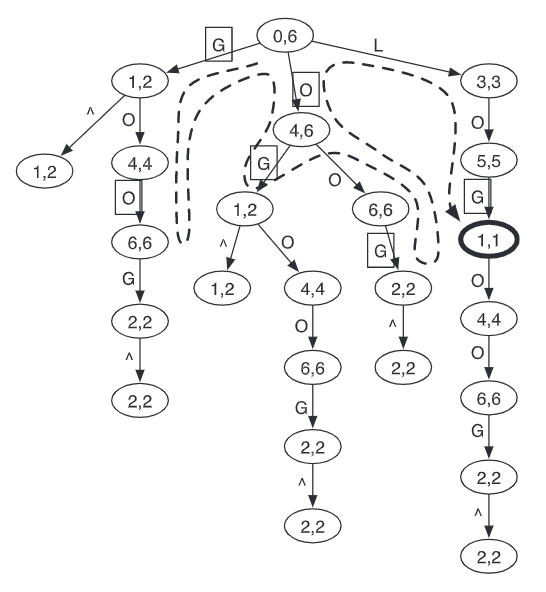
字符串X的前缀树中,每条边代表一个字符,每条从叶子节点到根节点的路径上的边上字符形成的字符串都是X的前缀,每个节点到根节点中的路径上的字符形成的字符串都是X的字串
Burrows–Wheeler transform
X表示字符串,X[i]表示字符串中的第i个字符,X[n-1]= $
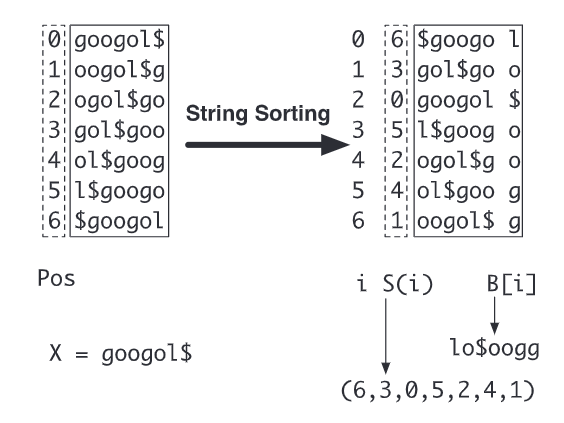
- 上图左边的数组是suffix array,用 $X_i$ 表示,$X_0$='googol$$$',$X_1$='oogol$$$'
- 上图右边的数组是经过字典排序后的suffix array
- S[i]: 经过字典排序后的第i个字符串在排序前(suffix array)的位置
- B[i]: 经过字典排序后的第i个字符串的最后一个字符
Burrows–Wheeler transform 转换后的结果是S和B
Suffix array interval and sequence alignment
对于一条字符串,SA interval 就是这条字符串在 经过字典排序后的suffix array 中作为子串的位置区间,SA interval 就是 prifix trie 中节点上的数字
例如:
gol只在字典排序后的suffix array位置为1的字符串中作为前缀,它所对应的就是[1,1]o在字典排序后的suffix array位置为4,5,6的字符串中作为前缀,它所对应的就是[4,6]
对于精确的匹配问题,只有一个解;对于不精确的问题,可能会有多个解
Exact matching: backward search
- a:字符表中的一个字符
- C(a):在X[0,n-2]中小于a(字典排序在a前面)的字符的数量
- O(a,i):在B[0:i]中a出现的次数
W是X的一个字串,$\underline R(aW)=C(a)+O(a,\underline R(W)-1)+1$ , $\overline R(aW)=C(a)+O(a,\overline R(W)$ ,当且仅当$\underline R(aW) \le \overline R(aw)$ 时,aW是X的字串
The fragment assembly string graph
伪码我在这里就不贴了,感兴趣可以去读原文.
BUILDING A STRING GRAPH
- 两个read的重叠时,重叠区的端点,在原read中有两个是read的端点
- 顶点 Vertic {type;read} type:B/E 表示为read的开始点或者结束点,read表示和这个顶点相关的read
- 每个read F是两个节点,一个节点时表示它的开始点F.B,一个表示他的结束点F.E
- 两个顶点之间相连是两个read的端点相连,一个端点处于重叠区中,另一个位于非重叠区
- 这样生成的图中有可以削减的边
- A->B->C和A->C,可以去掉A->C
- 如果有多个入度和出度都是1的节点相连,可以用一条复合边替代
- 任意两个有重叠区的read相连时,都会有两条边连接四个顶点,这两组是等价的,可以去掉一条边和两个顶点
A LINEAR TRANSITIVE REDUCTIONALGORITHM
Fast and accurate long-read assembly with wtdbg2
Main
-
使用的是基于overlap的方法,主要内容有:快速的all vs all read alignment 和 一种基于 fuzzy-Bruijin graph 的布局算法
-
contigs:
- read中256个base作为一个bin,在所有read的所有的bins之间进行 all vs all read alignment 和 构建 fuzzy-Bruijin graph
- binning 操作是为了加速计算
- fuzzy-Bruijin graph :一个vertex是four-bins,如果两个vertex在一个read中,就有一条边连接到这两个vertex
-
FDB:
- 类比:FBG中256base作为一个bin类似于DBG中的一个base,相应的K-mer变成K-bins,由k个连续的bins组成
Method
The wtdbg2 algorithm
- 过滤掉只出现一次或超过1000次的k-mer,为剩余的k-mer构建一个hashtable
- 遍历所有read,找到那些之间有共同k-mer的read,把每个bin作为一个base,在binned序列,gap 和不匹配的bins间使用Smith–Waterman-like dynamic programming ,保留那些不少于8*256bp的alignment
- 如果有两个k-bins,他们之间的overlap是k个bins,这两个就是等价的,我们把构造一个关于k-bins 的不冗余集合(里面所有的k-bins都是不等价的,且k-bin的数量最大),记录k-bins的位置和覆盖率
- 构建V,以覆盖率从大到小遍历集合中的k-bins,如果一个k-bin和V中有overlap的k-bin数量大于等于3或者大于这个新k-bins覆盖率的一半,把这个k-bin加入到V中,否则忽略
- 构建E,如果两个k-bins在一个read上,就在这两个k-bins间加一条边,因为两个k-bin可能会同时出现在不同的read上,所有会有不同的边,我们只保留一条边,并且计数,去除计数小于3的边
conclusion
- 主要就是拓展DBG吧,因为是长read而且有noisy,把256个base看作一个base,可以有效忽略噪声,还能加速。
Canu: scalable and accurate long-read assembly viaadaptivek-mer weighting and repeat separation
Methods
MinHash overlapping
- refer Assembling large genomes with single-molecule sequencing and locality-sensitive hashing
- 使用MHAP计算所有read之间的overlap,他有两个阶段,第一个阶段找到那些有可能有overlap的一对,
- TF-IDF算法 :TF(词频) x IDF(逆文档排序) ,简单快速容易理解,但重要性不够全面
- 第一阶段用了IF-IDF来加权k-mer,第二阶段用bottom sketch多重hash
Parallel overlap sort and index
- bucket sort
Read correction
LoRDEC: accurate and efficient long read error correction
Introduction
Related works for PacBio reads
- 测序错误在read中是均匀分布的,独立于序列的内容,并倾向于插入,缺失的较少
Contribution
- 本文提出了一种高效的复合纠错方法
- 首先构建SR的DBG,在DBG内搜索最佳路径来纠正LR