
MECAT: fast mapping, error correction, and de novo assembly for single-molecule sequencing reads
仅代表个人理解,如果有不准确的欢迎指正
可能会用到的知识点
- SMS:single-molecule sequencing 单分子测序技术
- mapping:只提供read比对的参考序列上的位置信息;alignment:不仅提供位置信息,还有他们的实际位点和参考位点之间的匹配、不匹配、插入或删除的关系
- global alignment :需要比对整个查询序列,查询序列在参考序列上的比对结果可能会不连续,一般用来比较同源基因,或者基因的差异性
- local alignment :查找最能匹配上参考序列的局部区域,比对上的结果可能是查询序列的子序列,
- Chimeric reads : occur when one sequencing read aligns to two distinct portions of the genome with little or no overlap. Chimeric reads are indicative of structural variation. Chimeric reads are also called split reads.
- contig : 重叠的read,根据read之间公共的片段形成
- DAG: Directed Acyclic Graph 有向无环图,图中从任意顶点出发无法经过若干条边回到该点
- DAGCon:
Main
SMS read 的 alignment 是非常耗时的,通常基于k-mer的方法是找种子(read 中的一段) 进行read间的alignment,但由于生物学基因的的重复性,位于重复区的read的k-mer能够匹配上的数量非常高,这会导致有额外的候选者,忽略高重复率的k-mer可能导致缺失正确的overlap,BLASR中是利用稀疏的动态规划来完成的,最近,canu中使用tf-idf k-mer weighting 方法来减少高重复k-mer的影响,但canu没有考虑k-mer之间的排列。
本文中的方法是使用距离影响因子DDF(distance difference factors)给k-mer对打分,包括所有的匹配k-mer对和他们的内部距离,因此,这个分数可以表示read之间的全局匹配信息,通过提取高分数的read pair来过滤不合适的候选者
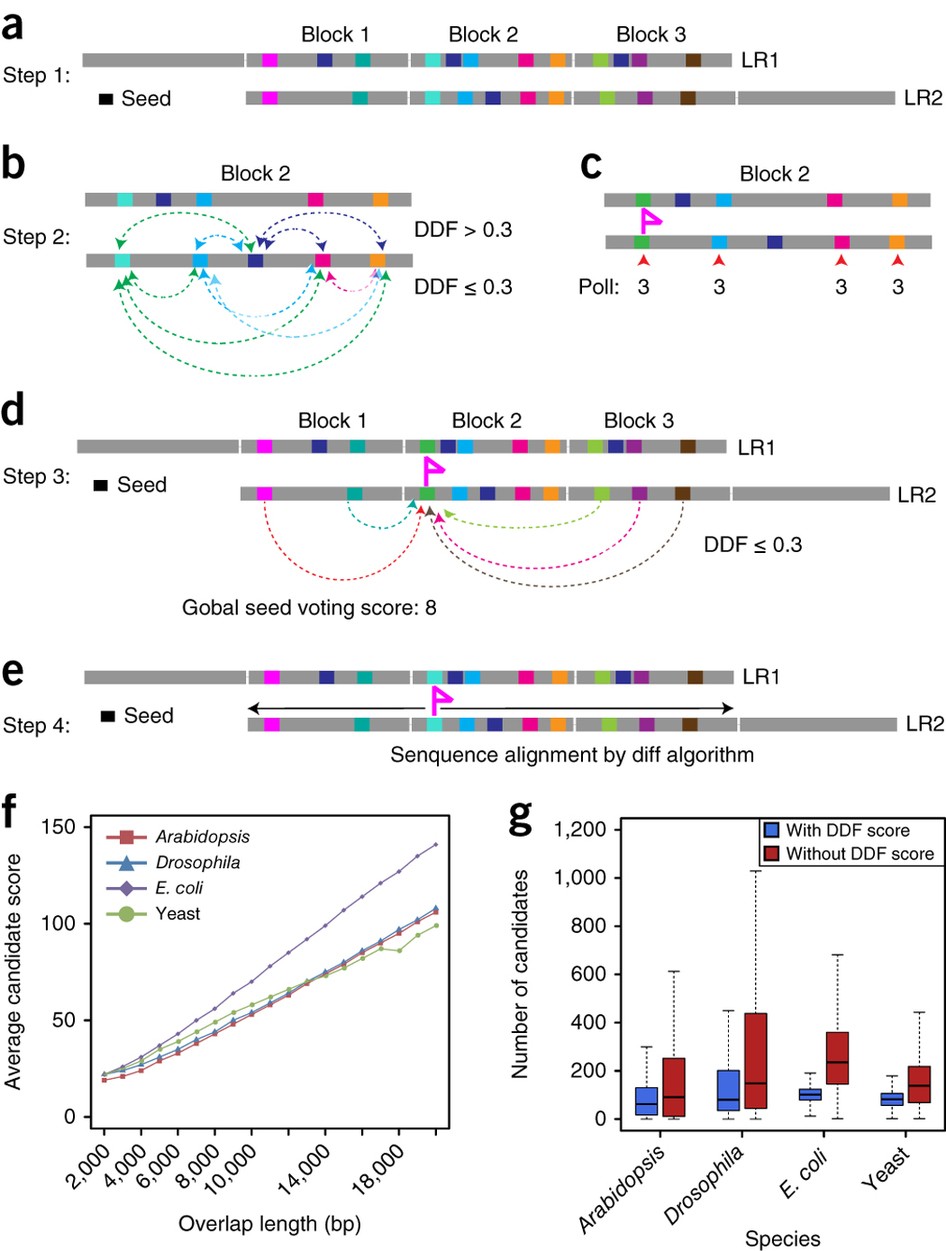
- a 两个read上block间的 k-mer alignment
- b 在block对里面,使用DDF对k-mer对打分,
- c 选择分数最高的作为k-mer种子,有分数相同的就随机选,(彩旗表示)
- d 加上其他block的k-mer对,对k-mer种子打分
- e 进行 sequence alignment
基于DDF score 算法,本文提供了 MECAT ,一个快速的比对工具,运行时可以选择是否使用 local alignment 。我们可以仅根据DDF分数来选择可靠的read对,当只需要候选匹配对时,我们可以不进行local alignment 。
DDF alignment score 对 overlap 长度敏感,所以MECAT 也适合 read到参考基因上的比对
由于SMS read的高错误率,必须要进行纠错,纠错后的read一般是根据一定数量匹配read的共同部分进行构建的,MECAT可以不使用local alignment 快速选择候选read
Methods
自己暂时只用 correct 和 assembly
template read 自己预先给定的 read
Correcting single-molecule sequencing reads.
一般来说,纠错有两个步骤,第一步是在成对的read之间查找overlap,第二步是根据相关 alignment 结果中的一致(consensus)部分构建正确的结果
在第一步中,我们使用不带 local alignment 的 MECAT 查找 overlap , 输出放在多个文件中,每个文件包括200000个read 的 overlap 匹配信息,对于每个 read 的匹配信息,我们根据DDF score 降序排序,我们在read和相匹配的read间,从DDF score最高的开始,进行 local alignment,为消除chimeric(嵌合) read和重复子序列的影响,过滤掉 overlap 长度小于 read pair 中长度较短的 read 90% 长度的 alignment 结果,一旦收到到了100个 overlaps 或者 align 了所有 read 就停止 local alignment。因为避免了计算不相关的overlaps可以加快alignment的时间。
第二步中,为了提高准确性和保持高效性,提出了结合 DAGCon and FalconSense 概念的一种新的适应性的 SMS read 纠错方法。总结了每对的比对结果,使用每个 base 上匹配,插入、删除的 read 数量构建了一个 consensus table
match_count,碱基位置能够相匹配的read的数量,insertion、deletion 同理
- 认定为匹配条件: match_count/(match_count + deletion_count) > 0.8 and insertion_count < 6
- 认定为删除条件: deletion_count/(match_count + deletion_count) > 0.8 and insertion_count < 6
- 对于更复杂的插入的情况,构建了一个局部 POG 并使用了动态规划来解决,因为复杂区域的一般来说小于 10 个 base ,通过小的 POG 可以很快的找到 consensus 具体细节见下方 Supplementary
第二步中,再模板read 和相匹配的 read 间进行 local alignment 时,需要随机读取储存的read,DAGCon 和 FalconSense 把 read 放到硬盘中,不支持随机读取。为了加速纠错过程,把所有的read都加载到内存中了
De novo assembly using single-molecule sequencing reads.
使用 SMS read 组装基因组有三步:找到 reads 和 template reads 间的 overlap,对 template reads 纠错,使用纠错后的 reads 构建 contigs。本文通过整合 canu 和新的 alignment 和纠错方法开发了两个新的pipeline。在 MECAT pipeline 的第一步,对所有超过3000bp的read运行分别进行alignment,从匹配上的read中选择分数最高的100个,在找overlap的过程中,不进行local alignment 使用mapping 信息进行纠错,第二步,用能够匹配到 template reads (>3000bp)的 read 对 template reads 纠错,最后,把纠错后的 read 使用 MECAT 中的 alignment 工具进行 alignment,然后把这些 alignment 结果放到 Canu 的 Unitig Construction 模型中去构建 unitigs 。或者直接把纠错后的read放到canu里。
Supplementary
DAGCon , FC_consensus 和 FalconSense是三个被广泛使用的 consensus 算法。他们有以下步骤:
- 根据两个 read 间的 alignment 构建一个多重序列的 alignment (multiple sequence alignment, MSA)
- 从多重 alignment 列中选择正确的碱基(base)
DAGCon 使用有向无环图将MSA编码成偏序 alignment (这应该和我之前代码的想法一致)。然而 FC_consensus 和 FalconSense 根据 base-level 的一致性和 偏序 alignment 使用标记和排序的方法构建一致性序列。
不太懂,等看了论文之后再重新整吧
DAGCon 有最高的准确性和最慢的速度(自己之后应该会选择这种方法,现在需要做的可能是找一个更准确的local alignment 工具,minimap2 的结果准确率只有95%,还是有点低,导致生成的一致性序列错误率很高,这个虽然可以通过多重纠错来解决,不过这就更麻烦了)
在研究中,提出了一个结合 DAGCon 和 FalconSense 概念的一个新的算法:在不重要的区域上,使用和 FalconSense 一样的基于计数的纠错方法,但在复杂区域(删除区域)通过 DAGCon 算法处理,但复杂区域一般都小于10个 base , 所以可以处理的很快
MECAT2CNS
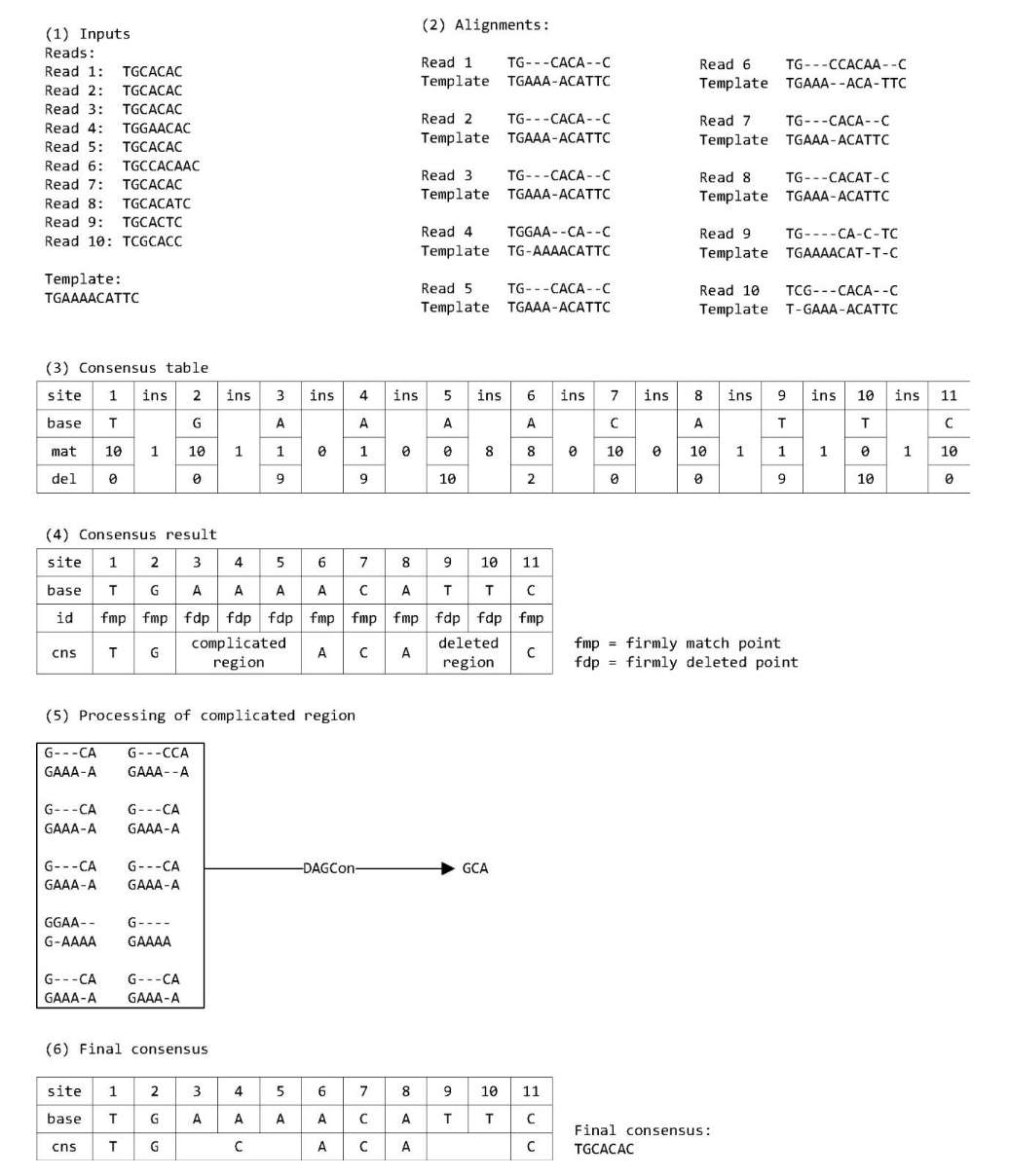
程序输入包含一个模板 read 和一组其他 read
- 构建一个 consensus table ,每个 base 都有匹配 mat 、插入 ins 、删除 del 三项记录表,分别记录匹配的个数,和在这个碱基后插入和删除的个数。
- 把所有 read align 到模板read上,如果首尾不匹配,可以截断头或尾以提高准确性
- 根据 alignment 的结果在表中填入相应的值
- 删掉不重要的计数 : mat + del < c ; ins < c
- 对于每列中
- mat/(mat + del) > 0.8 认定为 firmly matched point fmp ;
- del/(mat + del) > 0.8 认定为 firmly deletion point fdp ;
- 否则为不确定的点 underdetermined point fp
- 扫描整个 table,
- 如果是 fmp ,添加到 consensus 序列中,然后处理下一个
- 如果不是,继续扫描知道找到下一个 fmp ,如果这个之间只有 fdp,没有插入,则去除这段区域,否则定义为复杂区域,然后使用 DAGCon 算法处理,输出添加到一致性序列中,然后继续扫描。
本论文的大致写作思路
- 先介绍背景,三代测序数据。
- 介绍前人的工作结果和进展,部分会分析或者指出不足的地方
- 然后引出本文的主要内容方法和做出的成果,并且进行成果分析,以及和其他现有工具对结果进行比较
- 然后就是方法部分,分别就本文中的各个部分较为详细的介绍一一下自己的思路,具体的细节可能会放到 Supplementary 中说明