
Phased diploid genome assembly with single-molecule real-time sequencing
仅代表个人理解,如果有不准确的欢迎指正
可能会用到的知识点
- 等位基因!!!!!!
- SNP 单核苷酸多态性 SingleNucleotide Polymorphism 例如,对于某种生物,同一位置基因组片段一部分为
AAGC CTA,另一部分为AAGC T TA,则认为此处存在SNP、两种基因型属于等位基因。 - VS 结构体变异 Structural variation 生物染色体上结构的变异,多指涉长度介于1Kb与3Mb之间的序列大于单核苷酸多态性并且小于染色体畸变
Main
单分子实时测序技术 Single-molecule real-time (SMRT) 可以为哺乳动物提供高连续性的组装,long read 可以横跨多个重复区域,和帮助解决复杂的双倍体基因,本文提供了 FALCON 一个双倍体敏感的 long read 组装工具,FALCON-Unzip 一个相关的单倍体解析工具,用正确的同源染色体来组装代表具有 正确 定相 同源染色体的二倍体基因组的单倍体 contigs
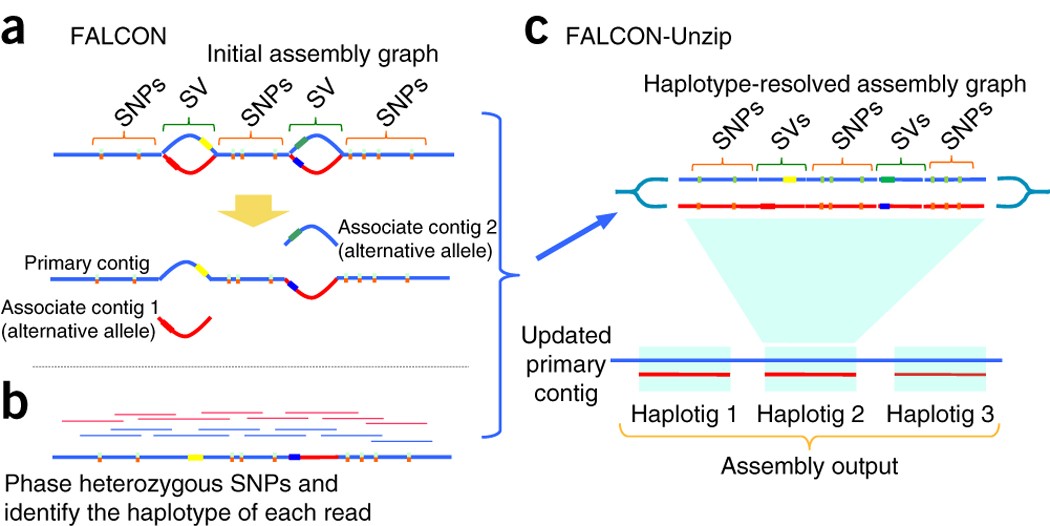
(a) 使用 FALCON 的一个最初的组装结果,先对原始 reads 纠错,然后通过关于 overlap 的 string graph 组装,组装后的 contigs 通过 FALCON-Unzip 完善成最后的 contigs 和 haplotigs
(b) Phase heterozygous SNPs and group reads by haplotype.
(c) The phased reads are used to open up the haplotype-fused path and generate as output a set of primary contigs and associated haplotigs.
FALCON assembler 首先用 reads 构建了包含单倍体融合(haplotype-fused)的 contigs和同源序列不同区域气泡的 string graph (a)。
FALCON-Unzip 使用来自其识别的杂合位置的定相信息识别读取单倍型(b)。
Method
Raw long-read error correction
使用 daligner 把所有的 read 都 align 到任意其他 read,这些 overlap 和 read 信息用 FALCON-sense 于生成 consensus 序列。FALCON-sense 算法可以保留杂合 SNP 的信息
Supplementary information
Updated FALCON consensus algorithm
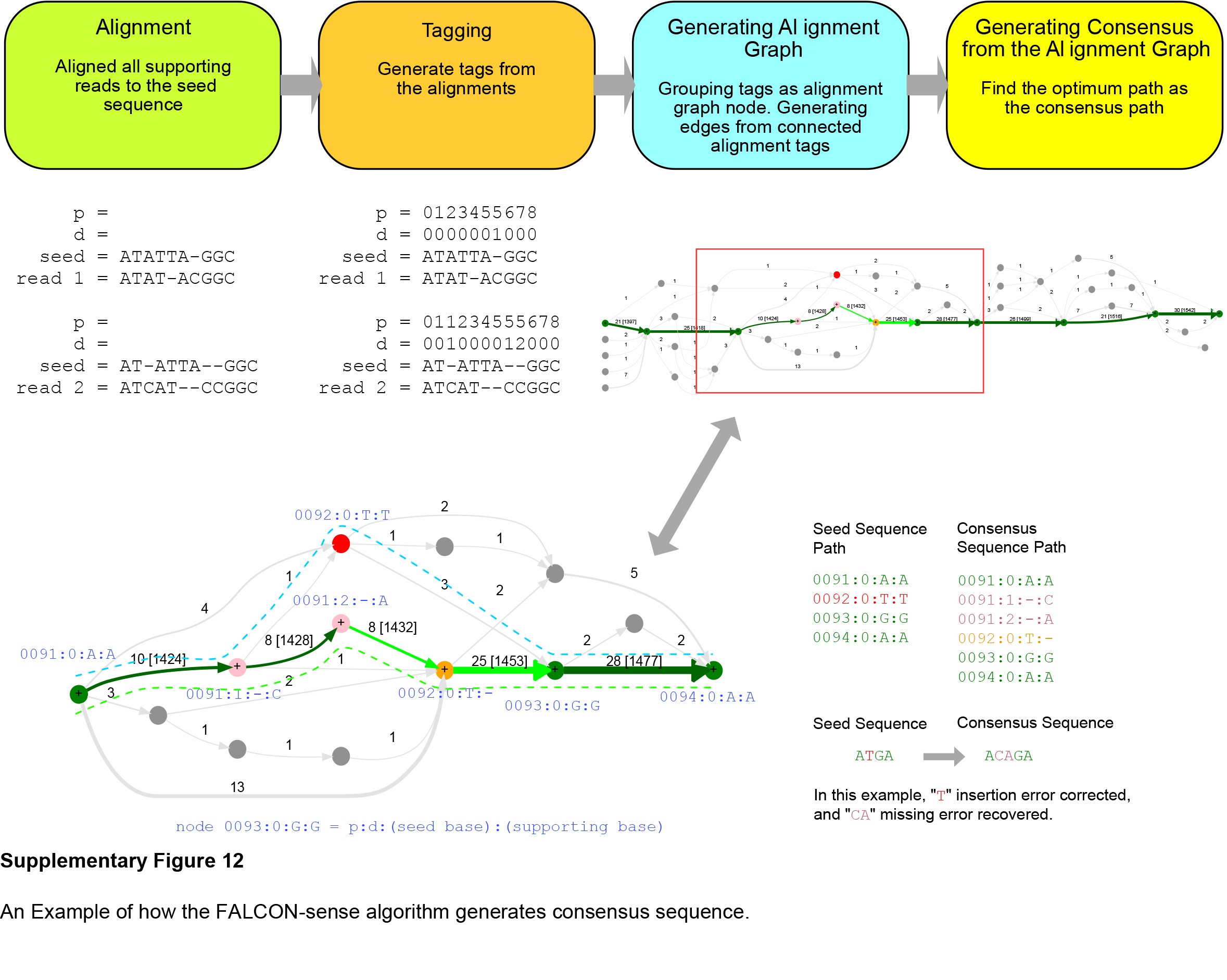
图中虚线突出显示了两条路径,青色虚线表示种子序列的路径,绿色虚线表示最后的 consensus 路径,Node “0092:0:T:T” 被认为是种子 read 上错误的额外碱基。新 Nodes 0091:0:-:C” and “0091:0:-:A” 添加到 consensus 路径,绿色箭头突出了原始的模板序列,插入到种子序列的 “CA” 与原始模板一致,也就是说,consensus 纠正了3个种子 read 中模拟的错误
FALCON-sense 使用一系列种子序列和支持他们的 reads 产生每个种子序列的 consensus
- 使用 Gene Myers' O(ND) aligner 把支持种子序列的 reads align 到种子序列上,在 alignment 中只允许插入和删除,不允许存在错配
- 在 alignment 的每一个位置都生成一个 alignment tag,tag 包含4个方面
- 在种子序列上的锚点位置
- 种子序列上锚点位置的 delta (插入的base数量)
- 种子序列上的base(碱基)
- alignment 中支持序列上的base
当在给定位置上的支持read相对于种子序列有插入时,alignment 上插入的 base 的非零 delta 表示来自锚点位置的 base 数量。tag 1 和 3 有相同的信息,只是表示不同,加入3只是为了方便
- 根据支持reads的 tag 构建一个 alignment graph
- 对于每个不同的tag,构建一个相应的node
- 对于 支持 read 上连续的tag,在图上创建一条边并给这条边上的 edge_count 赋值 1,如果这条边之前存在则 edge_count 的值 +1 ,结果是一个单项无环图(DAG),每一个 node 都有 tag,edge_count 表明有多少个read支持两个 nodes 之间的链接,edge_count 值越大表明支持 reads 上 nodes 之间的连接越可靠,一个高质量的 consensus sequence 也就是通过这些高可靠性的 edge 连接起来的 nodes 形成的一条链
- 在 alignment graph 上使用标准的动态规划算法找到最可靠的路径,通过串联路径上 tags 上的 bases 产生 consensus sequence
Assembly string graph reduction process summary
Constructing the initial string graph
在纠正完 reads 后,使用 daligner 获取 reads 间的 overlap。然后 overlap-filtering 步骤过滤掉已包含的 reads 和出现在高度重复区域的 reads。使用 Myers’ paper (Myers 2005)上的方法,根据过滤后的 reads pairs 构建 string graph。 本文通过 overlap 构建普通的单向图,每对 overlap 实际上构建了两条有向的边, 但是我们只需要沿着一个方向延伸,以获取 contigs 和它们反向互补相对应的部分。也就是说,在最终的 contigs 中只有每对有向边的一条。在构建结构阶段,我们根据每对边的对称性去除冗余。.
A heuristic algorithm for constructing compound paths 启发式算法构建复合路径
概念上讲,bubble 表示子图,就像(1)只有一个源节点和一个宿节点,(2)源节点到宿节点有多条路径,(3)在去掉任意一条边后,都保持着弱连接。复杂的杂合结构变异或二倍体数据集中两个单倍型之间的重复区域可能会导致更复杂的结构。
(1) 可能有嵌套 bubbles
_______
______/ \________
/ \_______/ \______
_____/ /
\_______________________/
(2) 图中也能有环
___<___
______/ \________
/ > \_______/ > \______
_____/ > /
\_______________________/
>
(3) 还有非常复杂的嵌套 bubbles,这可能表示某些局部复杂重复难以解决
______________
/ / \
/________/ \ / \
/____________\_/ \_______
____/___________________/ /
\______/ /__________/
\_____\___/
Falcon 用于寻找 bubbles 的复合路径的启发式方法如下所示:
- 把最初组装的 string graph 简化成一个具有简单路径的图。也即是说,把没有分支节点路径上的所有边变成一条边,把这个称为$UG_0$
- 对 $UG_0$ 中每个有多条出边的节点,搜索边的本地 bundle。设置 active tracers 追踪每个节点的每个分支
- 每次迭代都检查节点是否有 active tracers,如果一个节点的所有入节点都有一个 tracer ,给他的子节点添加 tracer 并且使它父节点的 tracer 变成 inactive,如果只剩一个 active tracer,那么多有遍历过的节点和边形成的子图称为复合路径(compound path)
- 如果任意一个active tracer 节点的后代有用过的 tracer,就表明探测到了一个循环,此时停止搜索并且不产生复合路径
- 基因复杂重复的部分,active tracer 会快速增加。限制 active tracer 的数量可以避免不必要的计算。如果数量超过了预设值,就停止搜索
- 对于每一步我们可以计算节点的数量和路径长度,路径长度可以看做从源节点开始到所有带有 active tracer 的节点的路径中有最多数量 bases 的路径。如果节点的数量或路径长度超过预设值,停止搜索
- 某些复合路径和其他路径之间有重叠部分,或者一个包含另一个,这样的小的 bubbles 是大 bubble 嵌入的一部分,这种情况下选择大的忽略小的
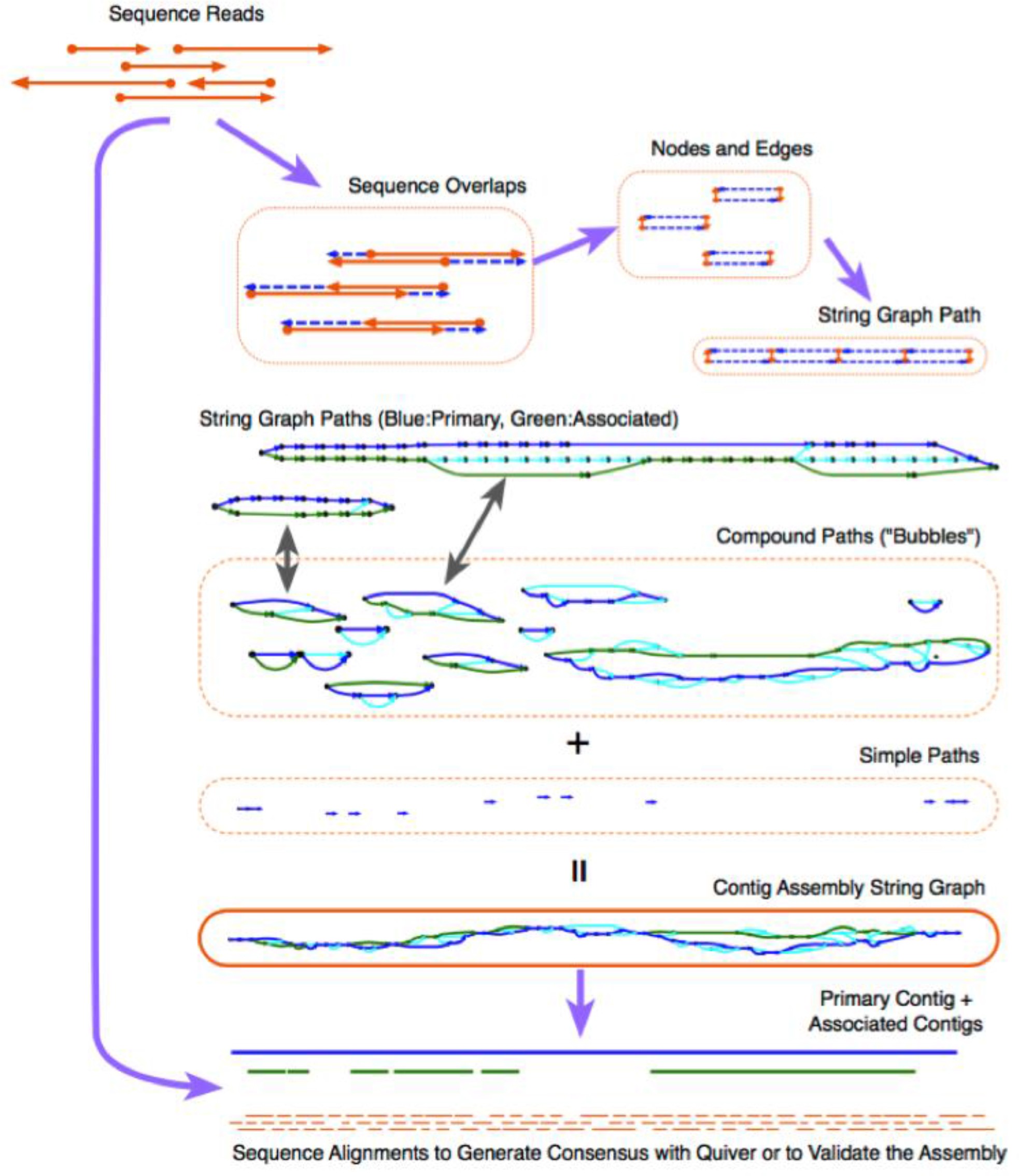
每个复合路径都是$UG_0$中简单路径的集合,$UG_0$中的简单路径可以被新确定的复合路径代替,可以形成一个联合图,图中的边要么是简单路径要么是复合路径。下一步是在联合图中确定没有分支的路径,和沿着边串联序列来产生 contigs。
From sequence overlaps to primary and associated contigs
上图总结了从纠错后的 read 通过多个图简化步骤到 contigs 的整个步骤。首先,把图分解成简单路径和复合路径,通过结合复合路径和简单路径构建出原始的 contig。这些 contig 仍然是单倍体混合的 contig。也就是说,不同区域的原始 contig 可能来自不同的单倍体,在这个阶段,布局规则不保证单倍体一致性,而且,两个单倍体之间有少部分不同时,不会有关联的 contig 生成。可以使用 ALCON-Unzip 去分离单倍体序列和生成单倍体相关的 contig。
A greedy SNPs phasing algorithm
构建单倍体序列,需要分离同源的 reads 到不同的组中,步骤如下:
-
查看 read 在原始 contig 中的位置,在 read 和 contig 之间生成 alignment,有两种策略
- 检查组装期间生成的 overlap 数据,确定属于每个特定 contig 的 reads 并把这些 reads 重新 align 到特定的 contig
- 简单的把 read align 到组装后的 contig
对于第一种,首先检查每个contig的平铺路径,检查完组装步骤生成的 overlap 数据后,每个 read 都可能和原始 overlap 数据中,分配到contig或未分配的有 overlap。分配给 contig 有 overlap 的 reads 可能不是来自同一个 contig。需要打分决定那个是最好的,通过 overlap 的长度给他们打分。如果有 overlap 最好的一对 read 被分配给了 contig,那么未分配的查询 read 也分配给这个 contig。把 reads 分组分成和原始 contig 相关的集合,每个集合和他们的 reads 分别独立的进行 align。策略1因为可以重新使用存在的 overlap 数据,可以节约算力
-
对每个 contig 调用 het-SNPs。对于大多数单倍体重构算法都需要 het-SNPs 结果作为输入,我们使用简单的 alignment 和计数方法来调用 het-SNPs。使用 BLASR 把 read 比对到 contig 上,生成 BAM/SAM 文件,只关注 SNP 并且忽略附近有插入或删除的 SNP ,也就是说只记录 BAM 文件 CIGAR 中的 “M” 模块。对于 contig 中的每个 base,记录 aligned read 每个 A、G、C、T 的数量,如果数量最高的小于75%,并且第二的大于25%,我们称之为一个 het-SNP位点,并且这两个 bases 作为 SNP 定相下游的两个等位基因 base
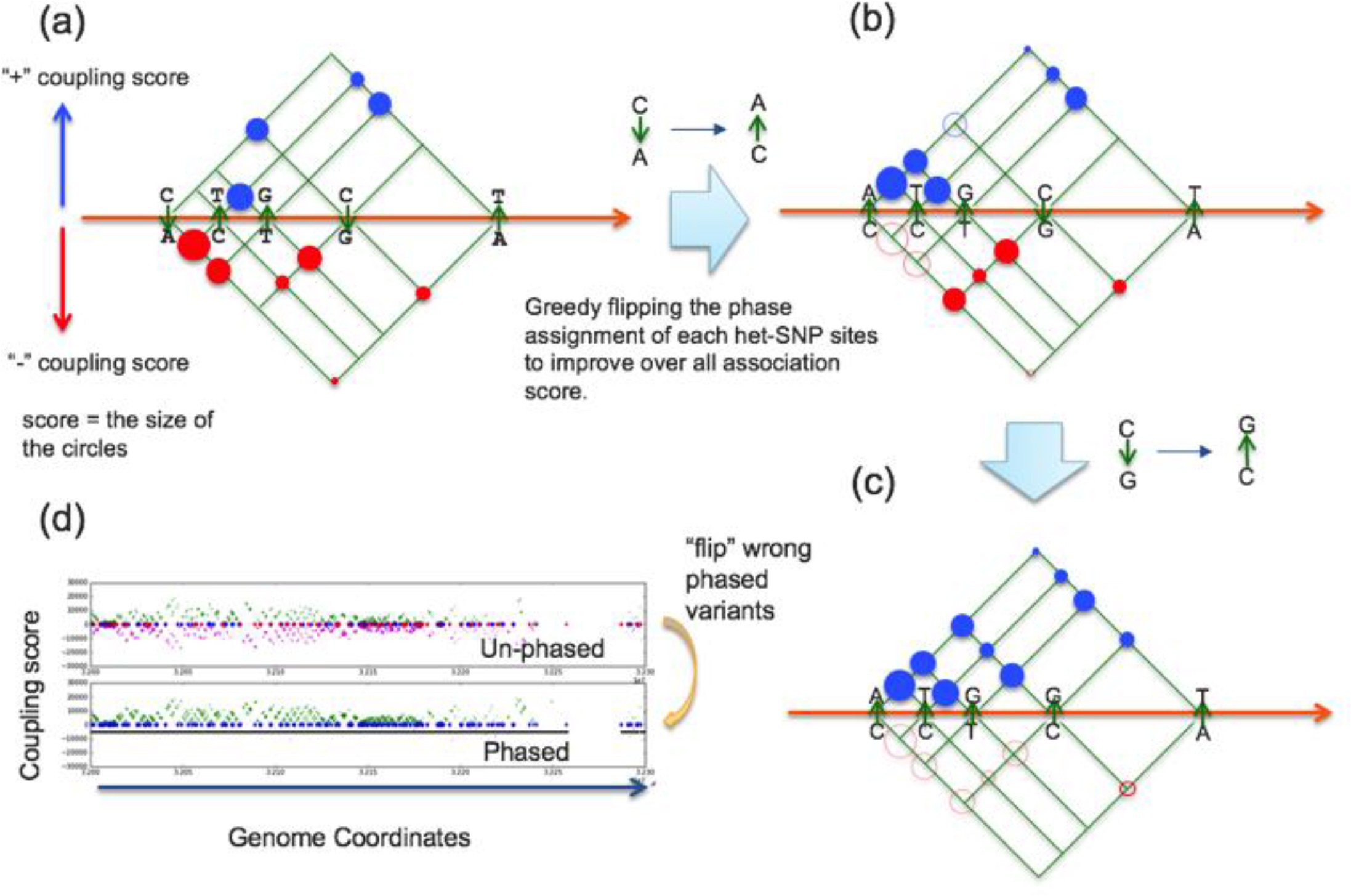
(a) All pairs of het-SNPs that are covered by multiple reads are evaluation. A “coupling score” is calculation from the number reads that support current haplotype assignment of the het-SNPs. (b)(c) We linearly scan through the het-SNP positions. If the total score is improved by flipping the haplotype assigned at one location, then we flip the assignment. (d) An example showing the “coupling score” before the flipping process (un-phased het-SNPs assignment) and afterward (phased het-SNP assignment).
-
对每个 het-SNPs 定向。使用简单的贪婪算法在不同的单倍体组中定相 SNPs ,一般模型如上图所示。For
each het-SNP site, the initial phase of the variants are randomly chosen. For any two sites
that have shared reads mapped on, we can calculate a “coupling score”. The “coupling
score” is defined simply as the number of reads supporting the particular phase. Along the
reference, we scan the het-SNP sites from the 5’-end to 3’-end. We test the two possible
choices of the phase at the given site for each het-SNP site. The choice with more “coupling
score” support is kept. When there are fewer than 6 reads connecting two het-SNP sites,
we break the haplotype blocks. We apply this scanning and updating process iteratively
until no more optimization of the score is possible or reach a pre-specified limit of
iterations. At the end of this process, the het-SNP sites along the reference will be grouped
into different haplotype blocks and we have a set of phased SNPs associated with each
block. -
Assign phases to the raw reads. For each read, we examine the alignment to the reference.
If the read contain het-SNP calls against the reference, we count which phases it agrees the
most and assign a tuple of “(block identifier, phase identifier)” to the read. For reads where
no haplotype block and phase assignment is possible, e.g. reads without enough het-SNP
calls, we simply assign a sentinel value for the block identifier to trace the un-phased reads.
The phasing method presented here is rather generic. It performs reasonably as shown in the
evaluation work presented in the main manuscript. We anticipate if we integrate more
sophisticate algorithms for phasing can further increase the accuracy and phasing contiguity.
Incorporate phased reads to haplotype-fused string graph for unzipping collapsed paths
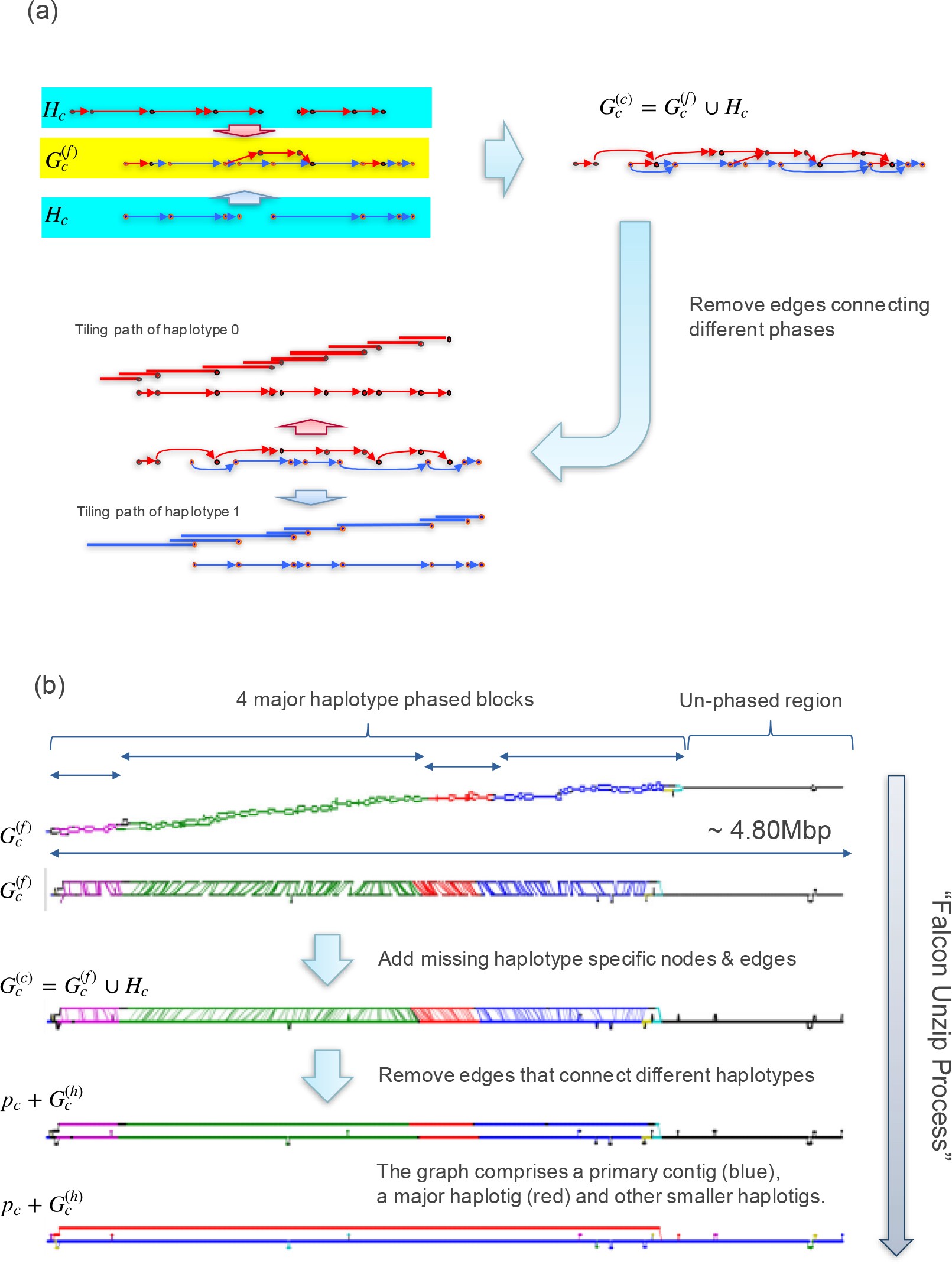
(a) shows the schematic of how to merge the haplotype specific
assembly graph Hc to the initial haplotype-fused graph Gc
(f)and reconstruct the haplotype
tiling paths for generating the haplotigs. The haplotype specific assembly graph has 4
disconnected subgraphs inside the cyan boxes, two for each haplotype. If we add the edges
and nodes from the Hc to the haplotype-fused graph in the yellow box, we get the
intermediate graph shown in the upper right panle. In the graph, the red nodes and blue nodes
indicate the reads from the two different haplotypes. There are some edges connecting the
nodes from two different haplotypes. Once the haplotype of the reads are known, we can
remove those edges. This results in the graph in lower panel. We can construct the tiling paths
for the two haplotigs from the two disconnected and haplotype specific components.
(b)shows an example constructing the primary contig and haplotigs
from the Clavicorona pyxidata assembly. Along the initial primary contig, we can find 4
haplotype blocks and an un-phased region. By applying the FALCON-Unzip process, we bring
back some reads to fully reconstruct the haplotigs. In this example, the four initial haplotype
block can be further merged by the overlaps across the bubbles in the assembly graph.